Universal Parasite Diagnostic (nUPDx): A Deep-Amplicon Sequencing Framework for High-Sensitivity Detection of Blood-Borne Parasites
This article explores the nested Universal Parasite Diagnostic (nUPDx), a targeted amplicon deep sequencing (TADS) approach that uses the 18S rDNA gene and selective restriction enzyme digestion to detect and...
Universal Parasite Diagnostic (nUPDx): A Deep-Amplicon Sequencing Framework for High-Sensitivity Detection of Blood-Borne Parasites
Abstract
This article explores the nested Universal Parasite Diagnostic (nUPDx), a targeted amplicon deep sequencing (TADS) approach that uses the 18S rDNA gene and selective restriction enzyme digestion to detect and differentiate a wide spectrum of blood-borne parasites with a sensitivity rivaling pathogen-specific qPCR. We detail the assay's evolution, including the Ad_UPDx modification that integrates library preparation and barcoding into PCR steps, drastically reducing cost and turnaround time. The content covers foundational principles, step-by-step methodology, troubleshooting for common NGS preparation issues, and rigorous validation against established diagnostics. Aimed at researchers and drug development professionals, this review synthesizes how nUPDx addresses critical gaps in conventional and molecular parasitology, offering a powerful tool for complex clinical cases, epidemiological surveillance, and veterinary diagnostics.
The Principles and Evolution of Universal Parasite Diagnostics
Accurate and timely diagnosis of parasitic and fungal infections remains a critical challenge in clinical and research settings. Conventional diagnostic methods, along with modern single-pathogen molecular tests, exhibit significant limitations that hinder effective patient management, disease surveillance, and drug development. These diagnostic gaps are particularly problematic for immunocompromised patients and in cases of co-infections, where delayed or inaccurate diagnosis can lead to severe outcomes [1]. The rising threat of fungal infections and the emergence of new parasitic pathogens highlight the urgent need for diagnostic approaches that can overcome the constraints of traditional methods.
The limitations of current diagnostic approaches span technical, operational, and economic dimensions. Microscopy, while cost-effective, suffers from variable sensitivity and operator dependence [2]. Single-pathogen molecular tests offer improved accuracy for targeted organisms but fail to detect unexpected or rare pathogens [3]. This application note examines these diagnostic gaps within the context of advancing universal parasite diagnostic (nUPDx) deep-amplicon sequencing research, providing researchers and drug development professionals with a comprehensive analysis of conventional limitations and the transformative potential of comprehensive sequencing approaches.
Comparative Performance of Diagnostic Methods
Quantitative Analysis of Diagnostic Limitations
Table 1: Performance Comparison of Diagnostic Methods for Parasitic and Fungal Infections
| Diagnostic Method | Sensitivity Limitations | Specificity Issues | Pathogen Coverage | Turnaround Time | Key Limitations |
|---|---|---|---|---|---|
| Microscopy | Variable (requires ~100 parasites/μL for reliable detection) [2] | Limited differentiation of related species [4] | Narrow; only visually distinctive pathogens | Minutes to hours | Operator-dependent; unable to differentiate colonization vs. infection [1] |
| Rapid Diagnostic Tests (RDTs) | Lower than molecular methods (92.4% vs. PCR for malaria) [2] | Cross-reactivity with related antigens [2] | Target-specific | 15-30 minutes | Cannot differentiate new vs. old infections [2] |
| Single-Pathogen Molecular Tests | High for targeted pathogens but zero for non-targeted | High for targeted pathogens | Extremely narrow | Hours to 2 days | Require prior suspicion of specific pathogen [3] |
| Culture-Based Methods | Suboptimal recovery (e.g., ~50% for Mucorales) [1] | Specific but prone to contamination | Limited to cultivable organisms | Days to weeks | Fragile hyphal structures may be damaged [1] |
| Nested TADS (nUPDx) | High (0.58 Plasmodium falciparum/μL) [5] | High with specific primer sets | Broad spectrum | 5 days [5] | Primer mismatches for some species [6] |
Table 2: Economic and Operational Considerations of Diagnostic Methods
| Method | Cost Per Sample | Equipment Requirements | Expertise Needed | Scalability | Adaptability to New Pathogens |
|---|---|---|---|---|---|
| Microscopy | $2.25-$3.40 [7] | Microscope, stains | High (experienced microscopist) | Low to moderate | None without new staining techniques |
| Single-Pathogen PCR | Varies by test | Thermocycler, DNA extraction system | Moderate | Moderate | Requires new primer/probe design |
| Multiplex PCR | $23.46 (MTBDRplus example) [7] | Specialized instrumentation | High | Moderate | Limited by panel design |
| nUPDx | ~$11 (modified assay) [5] | Illumina sequencer, bioinformatics | High (bioinformatics essential) | High | High with primer adjustments |
Technical and Operational Limitations in Practice
The deficiencies of conventional diagnostics extend beyond performance metrics to fundamental technical constraints. Microscopy-based identification of intestinal protozoa, while considered the reference standard in many settings, cannot differentiate between pathogenic and non-pathogenic species, such as distinguishing Entamoeba histolytica from non-pathogenic Entamoeba dispar [4]. This limitation has significant clinical implications, potentially leading to either unnecessary treatment or missed interventions.
Single-pathogen molecular tests address some specificity issues but introduce their own limitations. These tests require clinical presupposition of the causative agent, making them ineffective for detecting unexpected pathogens or co-infections [3]. Furthermore, the technical complexity of molecular methods, particularly those requiring specialized DNA extraction from robust parasitic cysts and oocysts, presents challenges for consistent performance across sample types [4]. The operational burden of these methods is substantial, with molecular assays like MTBDRplus demonstrating high labor requirements ($3.46 per test) and extensive laboratory facility needs [7].
For fungal diagnostics, the limitations are equally pronounced. Current methodologies for diagnosing Pneumocystis pneumonia face the challenge of differentiating colonization from active disease, while culture-based detection of Mucorales molds may fail in up to 50% of cases where hyphae are visible in stained specimens [1]. These gaps in fungal diagnostics are particularly concerning for immunocompromised patient populations, where delayed diagnosis significantly impacts outcomes.
Detailed Experimental Protocols
Universal Parasite Diagnostic (nUPDx) Deep-Amplicon Sequencing
The nUPDx protocol represents a significant advancement in comprehensive pathogen detection, enabling identification of multiple parasitic pathogens in a single assay [5] [3]. Below is the detailed experimental methodology:
Sample Preparation and DNA Extraction
- Collect 200-500μL of whole blood, tissue samples, or other biological specimens in EDTA or similar DNA-preserving collection tubes
- For tissue samples, homogenize using a gentle mechanical homogenizer to preserve DNA integrity
- Extract genomic DNA using magnetic bead-based nucleic acid purification systems (e.g., MagNA Pure 96 System)
- Include an internal extraction control to monitor extraction efficiency and potential inhibition
- Elute DNA in 50-100μL of TE buffer or molecular grade water
- Quantify DNA using fluorometric methods (e.g., Qubit dsDNA HS Assay)
18S rDNA Amplification with Modified Primers
- Prepare nested PCR reaction mixtures with primers incorporating Illumina barcodes and adapters
- First PCR reaction: Use universal eukaryotic 18S rDNA primers targeting variable regions
- Reaction composition: 1X PCR buffer, 2.5mM MgCl₂, 0.2mM dNTPs, 0.4μM each primer, 1.25U DNA polymerase, and 5μL template DNA in 25μL reaction volume
- Cycling conditions: Initial denaturation at 95°C for 5 min; 35 cycles of 95°C for 30s, 55°C for 30s, 72°C for 90s; final extension at 72°C for 7 min
- Second PCR reaction: Use nested primers with full Illumina adapter sequences for library preparation
- Purify amplicons using magnetic bead-based clean-up systems
Library Preparation and Sequencing
- Quantify purified amplicons using fluorometric methods
- Normalize concentrations to enable equimolar pooling of multiplexed samples
- Denature and dilute libraries according to Illumina MiSeq system specifications
- Load onto MiSeq flow cell targeting 50,000-100,000 reads per sample
- Perform 2x250bp or 2x300bp paired-end sequencing to ensure overlap of read ends
Bioinformatic Analysis
- Process raw sequencing data through quality control (FastQC)
- Merge paired-end reads (PEAR)
- Cluster sequences into operational taxonomic units (USEARCH, VSEARCH)
- Perform taxonomic assignment against curated 18S rDNA database (SILVA, NCBI)
- Analyze and visualize results (R packages: phyloseq, ggplot2)
Comparative Diagnostic Evaluation Protocol
To validate the performance of nUPDx against conventional methods, the following comparative protocol can be implemented:
Sample Collection and Processing
- Collect matched clinical samples (blood, tissue, stool) from patients with suspected parasitic infections
- Divide each sample into aliquots for parallel testing by different methods
- Process samples for microscopy: prepare thick and thin smears, stain with appropriate methods (Giemsa, calcofluor white)
- Preserve samples for molecular methods: freeze at -20°C or -80°C until DNA extraction
Parallel Diagnostic Testing
- Perform microscopy examination by experienced microscopists
- Conduct single-pathogen PCR assays for suspected pathogens
- Run commercial multiplex PCR panels if available
- Perform nUPDx deep-amplicon sequencing as described in section 3.1
- Include appropriate controls (positive, negative, extraction)
Data Analysis and Comparison
- Calculate sensitivity, specificity, positive predictive value, and negative predictive value for each method
- Use composite reference standard or latent class analysis for method comparison
- Identify discordant results and investigate via additional testing or clinical correlation
- Perform cost-effectiveness analysis comparing different diagnostic approaches
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagent Solutions for Universal Parasite Diagnostics
| Reagent/Category | Specific Examples | Function/Application | Performance Considerations |
|---|---|---|---|
| DNA Extraction Kits | MagNA Pure 96 DNA and Viral NA Small Volume Kit; QIAamp DNA Blood Mini Kit | Efficient lysis of diverse parasites; removal of PCR inhibitors | Critical for robust amplification; varies by parasite type and sample matrix [4] |
| 18S rDNA Primers | Modified universal eukaryotic primers with Illumina adapters [5] | Broad-range amplification of parasite 18S rRNA gene | Enables library prep without separate adapter ligation; coverage gaps for some species [6] |
| PCR Master Mixes | TaqMan Fast Universal PCR Master Mix; Hot-start high-fidelity polymerases | Efficient amplification with minimized nonspecific products | Critical for complex sample types; reduces background in sequencing |
| Library Prep Kits | Illumina DNA Prep; Nextera XT | Efficient library construction from amplicons | Impact sequencing efficiency; compatibility with amplicon size critical |
| Positive Controls | Synthetic gene fragments; characterized parasite DNA | Process monitoring; quantification standards | Essential for validating each run; should represent diverse parasite taxa |
| Bioinformatic Tools | DADA2; mmlong2; custom curation pipelines [5] [8] | Taxonomic assignment; quality control | Critical for accurate species identification; requires curated databases |
Visualizing Diagnostic Limitations and Solutions
The limitations of conventional and single-pathogen molecular tests create significant diagnostic gaps that impact clinical management, epidemiological surveillance, and drug development programs. Microscopy suffers from sensitivity limitations and operator dependence, while single-pathogen molecular tests cannot detect unexpected pathogens or co-infections. The universal parasite diagnostic (nUPDx) deep-amplicon sequencing approach represents a transformative solution that addresses these gaps through comprehensive pathogen detection, reduced turnaround time, and decreasing costs (approximately $11 per sample) [5].
Implementation of this advanced diagnostic methodology requires careful consideration of DNA extraction protocols, primer design, and bioinformatic analysis. The experimental protocols and reagent solutions outlined in this application note provide researchers and drug development professionals with the necessary framework to implement this technology, ultimately bridging the critical diagnostic gaps that hamper effective parasite and fungal infection management. As these comprehensive sequencing approaches continue to evolve, they hold the potential to revolutionize pathogen detection and usher in a new era of diagnostic precision in clinical and research settings.
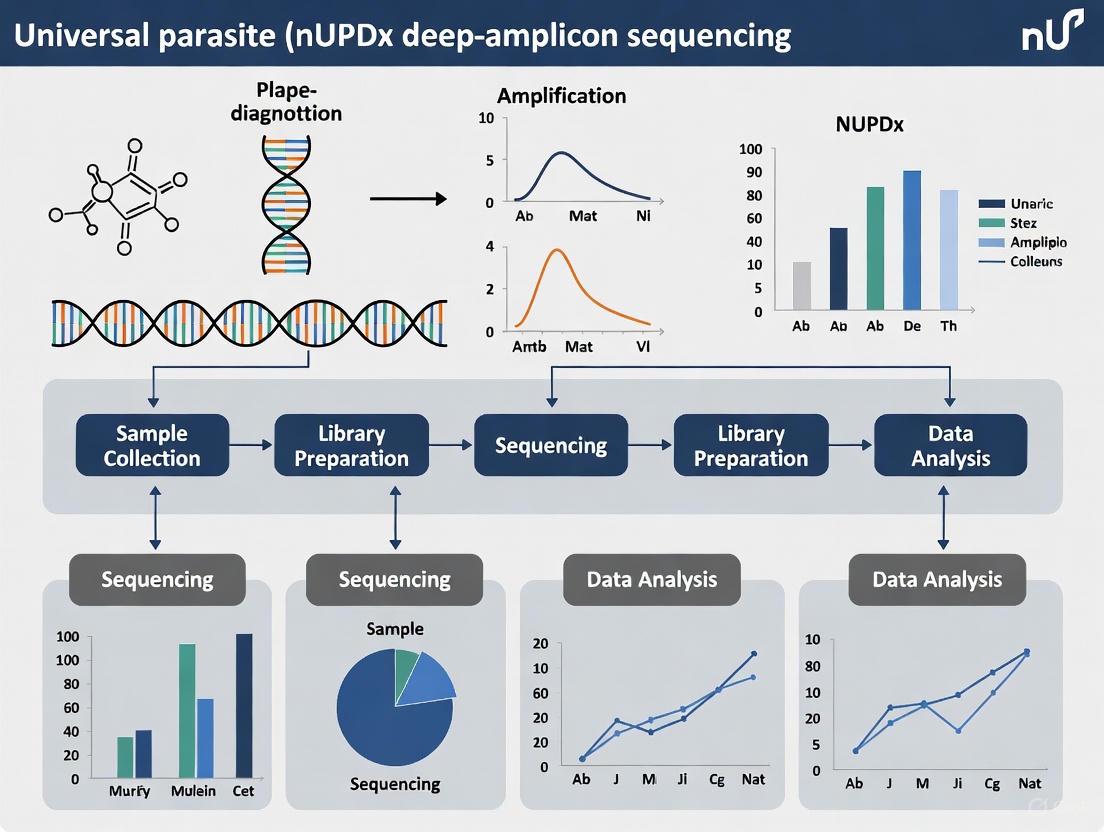
Targeted Amplicon Deep Sequencing (TADS) of the 18S rDNA locus represents a transformative molecular methodology designed to overcome the significant limitations of traditional parasite diagnostic techniques. This approach utilizes next-generation sequencing (NGS) of PCR-amplified regions of the highly conserved 18S ribosomal DNA (rDNA) gene to facilitate the universal detection and differentiation of diverse parasitic organisms. The 18S rDNA sequence contains a combination of conserved regions, which allow for the design of broad-range "pan-eukaryotic" primers, and variable regions, which provide the genetic diversity necessary for species-level identification [9]. Unlike traditional methods that require a priori knowledge of the suspected pathogen, this TADS-based universal parasite diagnostic (nUPDx) enables comprehensive screening for a wide spectrum of parasites in a single assay, proving particularly valuable for detecting unexpected, rare, or co-infections that might otherwise be missed by targeted methods [10] [3].
The core innovation of modern nUPDx tests lies in the strategic implementation of restriction enzyme digestion and a nested PCR workflow. This design selectively depletes abundant host-derived DNA in clinical samples, which has traditionally obscured parasite-derived sequences in metagenomic approaches [10]. By overcoming this fundamental challenge, the assay achieves a sensitivity comparable to, and in some cases exceeding, that of real-time PCR methods, with a documented limit of detection (LOD) for Plasmodium falciparum as low as 0.58 parasites/µL of blood [11]. This performance, combined with its broad diagnostic scope, makes 18S rDNA TADS a powerful tool for clinical diagnostics, veterinary medicine, and wildlife disease surveillance [3].
Performance Data and Key Applications
The performance of the 18S rDNA TADS assay is demonstrated by its high sensitivity and broad applicability across different sample types and parasite taxa. The following table summarizes key performance metrics and applications as validated in recent studies.
Table 1: Performance and Applications of 18S rDNA TADS for Parasite Detection
| Application / Parasite Group | Key Performance Findings | Sample Types Validated | Reference |
|---|---|---|---|
| Human Blood Parasites | LOD of 0.58 parasites/µL for P. falciparum; detects Plasmodium spp., Babesia spp., kinetoplastids, and filarial nematodes. | Human blood [11] [10] | |
| Universal Parasite Diagnostic (nUPDx) | ~10-fold lower LOD than previous TADS methods; sensitivity comparable to qPCR. | Human blood [10] | |
| Veterinary & Wildlife Diagnostics | Detected apicomplexan and nematode infections in mammals, birds, and reptiles; identified coinfections missed by microscopy/PCR. | Animal blood, tissues, whole parasites [3] | |
| Cost and Turnaround | Cost reduced to ~$11/sample; turnaround time shortened from 7 days to 5 days. | [11] | |
| Intestinal Protists (Metabarcoding) | Effectively detected and subtyped Blastocystis and archamoebid species from stool; lower sensitivity for flagellates like Giardia. | Human stool [12] |
A critical application of TADS extends beyond mere detection to the surveillance of antimalarial drug resistance [13] [14]. By targeting resistance genes such as Pfk13, Pfcrt, Pfmdr1, Pfdhfr, and Pfdhps, TADS enables high-throughput monitoring of known and emerging mutations within parasite populations. This approach provides a significant advantage over traditional capillary sequencing by accurately identifying low-frequency variants and complex mixed-genotype infections, which are crucial for early warning of resistance selection and spread [13]. For instance, a study in Kenya using TADS on dried blood spots confirmed the widespread presence of mutations conferring resistance to sulfadoxine-pyrimethamine while demonstrating the absence of artemisinin resistance markers at the time of the survey [13].
Experimental Protocol and Workflow
The following protocol details the nested TADS approach for the universal detection of blood parasites, which can be adapted for other sample types.
Sample Preparation and DNA Extraction
- Sample Collection: Collect blood specimens in EDTA tubes. Other sample types, such as tissues or stool, can also be used with appropriate processing [3] [12].
- DNA Extraction: Extract total DNA from samples using a commercial kit suitable for the sample type, such as the Fast DNA SPIN Kit for Soil or similar [15]. The extracted DNA contains a mixture of host and potential parasite DNA.
Nested PCR with Restriction Digestion
This streamlined workflow incorporates Illumina barcodes and adapters during PCR, eliminating the need for a separate, costly library preparation step [11].
First-Round PCR (Outer Primer Set):
- Primers: Use pan-eukaryotic primers (e.g., 1391F and EukBR) that flank the target ~200-bp region of the 18S rDNA gene. These primers include an overhang adapter sequence (e.g.,
TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGfor forward primer) for compatibility with the second PCR [15]. - Reaction Setup: Prepare PCR mix with high-fidelity DNA polymerase (e.g., KAPA HiFi HotStart ReadyMix), primers, and template DNA.
- Thermal Cycling:
- 95°C for 5 min (initial denaturation)
- 25-40 cycles of:
- 98°C for 30 s (denaturation)
- 55°C for 30 s (annealing)
- 72°C for 30 s (extension)
- 72°C for 5 min (final extension) [15].
- Primers: Use pan-eukaryotic primers (e.g., 1391F and EukBR) that flank the target ~200-bp region of the 18S rDNA gene. These primers include an overhang adapter sequence (e.g.,
First Restriction Digestion (D1):
- Following the first PCR, perform a restriction enzyme digestion on the product using PstI. This enzyme targets a cut site present within the human 18S rDNA amplicon but absent in the target parasites, thereby selectively reducing amplifiable host DNA [10].
Second-Round PCR (Inner Primer Set):
- Primers: Use inner pan-eukaryotic primers that are specific to the target ~200-bp region. These primers are fully tailed with the complete Illumina adapter sequences, indices (barcodes), and the target-specific sequence.
- Function: This step simultaneously amplifies the target region from the first PCR and adds all necessary sequences for Illumina sequencing, making the amplicons "sequencing-ready" [11].
- Thermal Cycling: Use a protocol similar to the first round, but with 30 amplification cycles [11].
Second Restriction Digestion (D2):
Pooling and Cleanup: Purify the final digested PCR products, quantify them, and pool equimolar amounts of each barcoded library for sequencing.
Sequencing and Bioinformatics Analysis
- Sequencing Platform: Sequence the pooled library on an Illumina MiSeq system using a v2 or v3 reagent kit (e.g., 2x150 bp or 2x250 bp paired-end reads) [11] [15].
- Bioinformatic Analysis:
- Demultiplexing: Assign reads to samples based on their unique barcodes.
- Quality Filtering & Denoising: Use tools like DADA2 or QIIME 2 to trim primers, filter low-quality reads, and correct sequencing errors to generate exact amplicon sequence variants (ASVs) [15].
- Taxonomic Assignment: Compare the resulting ASVs against reference databases (e.g., NCBI nucleotide database, custom parasite 18S rDNA databases) to assign taxonomic identities [15].
The following diagram illustrates the key steps of this protocol.
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of the 18S rDNA TADS protocol requires specific reagents and components. The table below details the essential materials and their functions.
Table 2: Key Research Reagents for 18S rDNA TADS Workflow
| Reagent / Component | Function / Role in the Protocol | Examples / Specifications |
|---|---|---|
| Pan-Eukaryotic Primers | Broadly amplify a hypervariable region of the 18S rDNA gene from diverse parasites while also containing sequences for Illumina adapters. | Outer primers (1391F/EukBR); Inner primers with full Illumina tails [10] [15]. |
| Restriction Enzymes | Selectively digest host-derived 18S rDNA amplicons based on cut sites present in vertebrates but absent in target parasites. | PstI (for D1); BamHI-HF and BsoBI (for D2) [11] [10]. |
| High-Fidelity PCR Mix | Ensures accurate amplification of the target region during multiple PCR cycles, minimizing introduction of polymerase errors. | KAPA HiFi HotStart ReadyMix [15]. |
| Illumina Sequencing Kit | Provides the chemistry for cluster generation and sequencing on the Illumina platform. | MiSeq Reagent Kit v2 or v3 [11] [15]. |
| Bioinformatics Tools | Process raw sequencing data, perform quality control, denoising, and assign taxonomic identity to sequences. | QIIME 2, DADA2, NCBI nucleotide database [15]. |
Critical Factors for Optimization
Several technical factors are crucial for optimizing the performance and accuracy of the 18S rDNA TADS assay:
Primer Specificity and Annealing Temperature: The design of pan-eukaryotic primers is critical for balanced amplification of different parasite taxa. Variations in the amplicon PCR annealing temperature can significantly affect the relative abundance of output reads for each parasite, potentially leading to biased representation [15]. Optimization of this parameter is necessary for a truly universal detection profile.
Impact of DNA Secondary Structure: The secondary structure of the 18S rDNA V9 region has been shown to have a negative association with the number of output reads for a given parasite species [15]. This molecular characteristic can create amplification biases where certain parasites are overrepresented while others are underrepresented in the final sequencing data, which must be considered when interpreting results.
Limitations in Protozoal Coverage: While the assay demonstrates high sensitivity for many parasites, it can have limited sensitivity for some common flagellates. Studies have reported failure to detect Giardia-specific reads in known positive samples and low sensitivity for Dientamoeba fragilis [12]. This highlights the importance of understanding the inherent limitations and primer mismatches that may affect detection of specific parasite groups.
Within the framework of universal parasite diagnostic (nUPDx) deep-amplicon sequencing research, a significant technical challenge is the overwhelming abundance of host DNA in clinical samples, which can obscure the detection of parasitic pathogens. The Host Depletion Strategy, which exploits vertebrate-specific restriction enzyme sites, is a targeted amplicon deep sequencing (TADS) method designed to overcome this limitation [10]. This approach enables the selective digestion of host-derived 18S rDNA, thereby enriching samples for parasite-specific sequences and significantly improving the sensitivity of parasite detection [16] [10]. The nUPDx assay, which incorporates this strategy, has demonstrated a limit of detection (LOD) comparable to pathogen-specific real-time PCR assays, facilitating the identification of major human blood parasites, including Plasmodium spp., Babesia spp., kinetoplastids, and filarial nematodes [16]. Recent advancements have streamlined this protocol, reducing both cost and turnaround time, thereby enhancing its potential for routine diagnostic applications [16]. This application note details the experimental protocols and key reagents essential for implementing this powerful host depletion strategy.
Core Principle and Workflow
The fundamental principle of this host depletion strategy lies in the bioinformatic identification of restriction enzyme cut sites that are present within the target 18S rDNA amplicon of vertebrates but are conspicuously absent in a broad range of blood-borne parasites [16] [10]. This taxonomic difference allows for the selective enzymatic digestion of host-derived DNA before and during a nested PCR amplification process, thereby proportionally enriching the sample for amplifiable parasite DNA [10].
The following workflow diagram illustrates the key steps in the nested PCR assay with integrated host depletion:
- Primary Restriction Digestion (D1): The total DNA extract, which contains a high proportion of host DNA, is first subjected to a restriction enzyme digestion. This initial digestion uses an enzyme (e.g., PstI) that targets a cut site within the host's 18S rDNA region, which is amplified by the outer primers [10]. This step digests a significant portion of the host DNA, reducing its capacity to be amplified in the subsequent PCR.
- First PCR (Outer Primers): The digested DNA is then amplified using a set of pan-eukaryotic outer primers. These primers flank the target region and are designed to amplify DNA from a wide range of eukaryotes, including both host and parasite [10].
- Secondary Restriction Digestion (D2): The product from the first PCR undergoes a second round of restriction digestion. This step utilizes one or more enzymes (e.g., BamHI-HF and BsoBI) that target cut sites found within the host's ~200-bp inner amplicon but not in the homologous sequences of target parasites [16] [10]. This further digests any host amplicons that were generated during the first PCR.
- Second PCR (Inner Primers): A nested PCR is performed on the doubly digested product using inner primers. These primers target the ~200-bp region within the larger amplicon generated in the first PCR [10]. The combination of two digestion steps and a nested PCR approach drastically reduces the proportion of host-derived reads and significantly enhances the sensitivity for detecting parasite DNA [16].
- Sequencing and Analysis: The final amplicons are sequenced on a high-throughput platform like the Illumina MiSeq. The resulting data is then analyzed bioinformatically to detect and differentiate parasitic species based on the 18S rDNA sequences [16].
Performance Metrics and Validation
The performance of the host depletion strategy has been rigorously validated using clinical samples and cultured parasites. The table below summarizes key quantitative data for the adapter-incorporating UPDx method (Ad_UPDx), an optimized version of the assay.
Table 1: Performance Metrics of the Ad_UPDx Assay
| Parameter | Original nUPDx Assay | Improved Ad_UPDx Assay | Experimental Context |
|---|---|---|---|
| Limit of Detection (LOD) | Comparable to conventional PCR [10] | 0.58 P. falciparum parasites/μL [16] | Determined using serially diluted, quantified cultures of P. falciparum spiked into parasite-free blood [16]. |
| Assay Turnaround Time | ~7 days [16] | ~5 days [16] | Time from sample processing to results. |
| Cost Per Sample | ~$40 USD [16] | ~$11 USD [16] | Includes reagents and sequencing. |
| Key Parasites Detected | Plasmodium spp., Babesia spp., kinetoplastids (Leishmania, Trypanosoma), filarial nematodes (Loa loa, Brugia malayi) [16] [10] | Plasmodium spp., Babesia spp., kinetoplastids, filarial nematodes [16] | Validated on clinical blood samples confirmed by PCR and/or microscopy [16]. |
| Application Scope | Human blood specimens [10] | Human blood; also successfully applied to blood, tissues, and other biological samples from mammals, birds, and reptiles [3] [6] | Demonstrated detection of apicomplexans, nematodes, and pentastomids in animal specimens [3]. |
Detailed Experimental Protocol
Sample Preparation and DNA Extraction
Materials:
- Clinical blood samples (e.g., collected in EDTA tubes).
- DNeasy Blood & Tissue Kit (Qiagen) or equivalent [17].
Procedure:
- Extract genomic DNA from 200 μL of whole blood using the DNeasy Blood & Tissue Kit, following the manufacturer's instructions [17].
- Elute the DNA in a final volume of 50-100 μL of Buffer AE or nuclease-free water.
- Quantify the DNA concentration using a fluorometer (e.g., Qubit). Store extracted DNA at -20 °C until use.
Primary Restriction Digestion (D1)
Materials:
- Restriction Enzyme: PstI (or an enzyme specific for the host's outer amplicon).
- Appropriate restriction enzyme buffer (e.g., NEBuffer).
- Nuclease-free water.
Reaction Setup:
- Total DNA Extract: Variable volume (e.g., up to 20 μL containing ~100-500 ng DNA).
- 10X Restriction Buffer: 5 μL.
- PstI Enzyme: 1 μL (10-20 units).
- Nuclease-free Water: to a final volume of 50 μL.
Procedure:
- Combine all components in a sterile microcentrifuge tube on ice.
- Mix gently by pipetting and centrifuge briefly.
- Incubate at 37 °C for 1 hour.
- Proceed directly to the First PCR or heat-inactivate the enzyme if required by the protocol.
First PCR (Outer Pan-Eukaryotic Amplification)
Materials:
- Pan-eukaryotic outer primers.
- High-fidelity DNA polymerase (e.g., Q5 Hot Start High-Fidelity DNA Polymerase, NEB).
- dNTPs.
Primer Sequences (Example):
- Forward Outer: 5'--[sequence as designed in original assay]-3'
- Reverse Outer: 5'--[sequence as designed in original assay]-3'
Reaction Setup:
- Digested DNA Template: 2-5 μL.
- 2X PCR Master Mix: 25 μL.
- Forward Outer Primer (10 μM): 1.25 μL.
- Reverse Outer Primer (10 μM): 1.25 μL.
- Nuclease-free Water: to 50 μL.
Thermocycling Conditions:
- Initial Denaturation: 98 °C for 30 seconds.
- Amplification (35 cycles):
- Denature: 98 °C for 10 seconds.
- Anneal: °C for 30 seconds.
- Extend: 72 °C for 30 seconds.
- Final Extension: 72 °C for 2 minutes.
- Hold: 4 °C.
Secondary Restriction Digestion (D2)
Materials:
Reaction Setup:
- First PCR Product: 20 μL.
- 10X Compatible Buffer: 5 μL.
- BamHI-HF Enzyme: 1 μL.
- BsoBI Enzyme: 1 μL.
- Nuclease-free Water: 23 μL (to a final volume of 50 μL).
Procedure:
- Combine all components in a sterile microcentrifuge tube.
- Mix gently and centrifuge briefly.
- Incubate at 37 °C for 1 hour.
- The digested product can be used directly in the next PCR or diluted 1:10 to reduce carryover inhibition.
Second PCR (Nested PCR with Adapter Incorporation)
Materials:
- Pan-eukaryotic inner primers with Illumina overhang adapters.
- Index primers (i7 and i5) for sample multiplexing.
- High-fidelity DNA polymerase.
Primer Sequences (Example with Adapters):
- Forward Inner: 5'--[Illumina Adapter + Overhang + Inner Forward Sequence]-3'
- Reverse Inner: 5'--[Illumina Adapter + Overhang + Inner Reverse Sequence]-3'
Reaction Setup:
- D2-Digested PCR Product (diluted): 2 μL.
- 2X PCR Master Mix: 25 μL.
- Forward Inner Primer (10 μM): 1.25 μL.
- Reverse Inner Primer (10 μM): 1.25 μL.
- Nuclease-free Water: to 50 μL.
Thermocycling Conditions:
- Initial Denaturation: 98 °C for 30 seconds.
- Amplification (15-25 cycles):
- Denature: 98 °C for 10 seconds.
- Anneal: [Y] °C for 30 seconds.
- Extend: 72 °C for 30 seconds.
- Final Extension: 72 °C for 2 minutes.
- Hold: 4 °C.
Library Purification and Sequencing
- Purify the final PCR product using magnetic beads (e.g., AMPure XP beads) to remove primers and enzyme.
- Quantify the purified library using a fluorometer.
- Pool equimolar amounts of each indexed library.
- Sequence the pooled library on an Illumina MiSeq system using a MiSeq v2 (500-cycle) or similar reagent kit, following the manufacturer's instructions [16].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of this host depletion strategy relies on a specific set of reagents and tools. The following table catalogs the essential components.
Table 2: Key Research Reagent Solutions for Host Depletion Assays
| Reagent Category | Specific Example | Function in the Workflow |
|---|---|---|
| Restriction Enzymes | PstI [10] | Primary digestion (D1); targets host sequence in outer amplicon. |
| Restriction Enzymes | BamHI-HF, BsoBI, XmaI [16] [10] | Secondary digestion (D2); targets host sequence in inner amplicon. |
| DNA Polymerase | Q5 Hot Start High-Fidelity DNA Polymerase (NEB) | High-fidelity amplification during nested PCR steps. |
| Pan-Eukaryotic Primers | Custom 18S rDNA primers (Outer & Inner sets) [16] [10] | Amplification of target region from a broad range of eukaryotic parasites. |
| Index Primers | Illumina i7 and i5 indexing primers [16] | Allows for multiplexing of samples during sequencing. |
| DNA Extraction Kit | DNeasy Blood & Tissue Kit (Qiagen) [17] | High-quality genomic DNA extraction from blood and tissues. |
| Library Purification | AMPure XP Beads (Beckman Coulter) | Purification and size selection of final sequencing libraries. |
| Sequencing System | Illumina MiSeq | High-throughput amplicon sequencing platform. |
| Host Depletion Kits (Alternative) | QIAamp DNA Microbiome Kit (Qiagen) [18] [19] | Commercial kit for microbial/environmental samples; uses enzymatic digestion different from the vertebrate-specific restriction method. |
| Host Depletion Kits (Alternative) | NEBNext Microbiome DNA Enrichment Kit (NEB) [18] [19] | Commercial kit for microbial/environmental samples; uses methylation-dependent digestion. |
The host depletion strategy that exploits vertebrate-specific restriction enzyme sites provides a robust, sensitive, and cost-effective foundation for universal parasite detection via deep-amplicon sequencing. By selectively digesting host DNA, the nUPDx assay and its derivatives overcome a major bottleneck in the metagenomic identification of eukaryotic pathogens. The detailed protocols and reagent specifications outlined in this document provide researchers with a clear roadmap for implementing this powerful diagnostic tool, which has demonstrated high sensitivity for detecting clinically relevant blood parasites and has been successfully adapted for use in veterinary and wildlife surveillance [3]. Continued refinement of primers, enzymes, and integration with novel sequencing technologies will further solidify the role of this strategy in modern parasitology and diagnostic test development.
The accurate diagnosis of parasitic infections presents a significant challenge in clinical and veterinary settings. Clinicians often face complex cases where a parasitic infection is suspected but difficult to pinpoint, requiring multiple pathogen-specific tests that demand a priori knowledge of the potential etiological agent [20]. To address this diagnostic limitation, researchers developed the Universal Parasite Diagnostic (UPDx) assay, a targeted amplicon deep sequencing (TADS) approach capable of detecting any parasite present in a clinical specimen using a single test [20] [21]. This innovative method employs pan-eukaryotic primers targeting the 18S rDNA gene, allowing theoretically universal detection of parasitic pathogens [10].
The initial UPDx assay demonstrated promise but faced a critical limitation: its sensitivity was only comparable to conventional PCR methods, rendering it less suitable for routine diagnostic applications where higher sensitivity is required [10]. The fundamental challenge stemmed from the overwhelming presence of host DNA in clinical specimens, which significantly reduced the proportional representation of parasite-derived sequences during sequencing [10]. This application note details the systematic enhancement of the UPDx assay through the introduction of a nested PCR approach with dual restriction enzyme digestion (nUPDx), resulting in substantially improved sensitivity while maintaining broad parasitic detection capabilities.
Technical Evolution: From UPDx to nUPDx
Fundamental Principles of the UPDx Assay
The original UPDx assay leveraged a clever molecular strategy to overcome host DNA interference. The assay targeted a specific ~200-bp region of the 18S rDNA gene that contained restriction enzyme cut sites present in vertebrates but absent in blood protozoa and filarial nematodes [10]. Specifically, the human 18S rDNA amplicon possessed BamHI-HF and XmaI restriction sites that could be exploited for selective digestion prior to PCR amplification [10]. This pre-PCR digestion step reduced amplifiable host DNA, consequently increasing the relative proportion of parasite-derived reads during subsequent sequencing. While this approach successfully reduced host-derived reads by more than 50% and increased parasite-derived reads by 5-10 times compared to undigested samples, its sensitivity remained limited, detecting only 0.58 Plasmodium falciparum parasites/μL of blood [5] [10].
The Enhanced nUPDx Design
The nUPDx assay introduced a nested PCR approach with dual restriction enzyme digestion to significantly enhance sensitivity [10]. Key improvements included:
- Extended Primer Design: A new set of pan-eukaryotic primers was designed with priming sites flanking the original ~200-bp target, enabling nested PCR amplification of the same locus.
- Dual Digestion Strategy: The nested approach incorporated two separate restriction enzyme digestions—one on the total DNA extract prior to the first PCR (D1), and a second on the product of the first PCR preceding the second PCR (D2).
- Enzyme Optimization: The second digestion (D2) utilized BsoBI restriction enzyme instead of XmaI, taking advantage of its cut sites within the human target amplicon that are absent in parasites [10].
This refined approach enabled a higher number of amplification cycles while progressively reducing host DNA contamination at two critical points in the workflow.
Table 1: Key Modifications from UPDx to nUPDx
| Feature | Original UPDx | Enhanced nUPDx |
|---|---|---|
| Amplification Strategy | Single PCR | Nested PCR |
| Restriction Digestion | Single digestion (pre-PCR) | Dual digestion (pre-PCR & between PCRs) |
| Digestion Enzymes | BamHI-HF & XmaI | PstI (D1) & BsoBI (D2) |
| Target Amplicon | ~200-bp 18S rDNA region | Same region with flanking primers |
| Sensitivity (LOD) | ~0.58 P. falciparum/μL [5] | ~0.58 P. falciparum/μL with 10-fold increased sensitivity [10] |
| Assay Turnaround | 7 days [5] | 5 days [5] |
| Cost per Sample | ~$40 [5] | ~$11 [5] |
Comparative Performance Data
Sensitivity Enhancements
The nUPDx assay demonstrated substantially improved performance characteristics compared to its predecessor. The critical advancement was its approximately 10-fold lower limit of detection (LOD), bringing it within the range of most qPCR methods [10]. This enhanced sensitivity was consistently demonstrated across multiple parasite genera, including Babesia, Plasmodium, various kinetoplastids, and filarial nematodes [10]. The assay maintained excellent specificity while detecting major human malaria parasites and other clinically important blood parasites [10].
Validation studies demonstrated the nUPDx assay's practical utility in diverse settings. When applied to 32 parasite-positive mammalian samples, the assay confirmed apicomplexan and/or nematode infections in 24 specimens, while additionally identifying several previously undetected coinfections [3]. The assay also detected infections in 6 of 13 positive bird samples and 1 of 2 positive reptile samples [3]. Importantly, the nUPDx assay identified Babesia sp. infections in 5 of 13 samples that had previously tested negative by other diagnostic approaches [3].
Operational Improvements
Beyond sensitivity enhancements, the nUPDx assay incorporated modifications that improved its practical implementation. The incorporation of Illumina barcodes and adapters directly during PCR eliminated the need for a separate library preparation step, reducing both turnaround time and costs [5]. These modifications decreased assay turnaround time from 7 days to 5 days and reduced the cost per sample from approximately $40 to $11, making the approach significantly more amenable to routine diagnostic applications [5].
Table 2: Detection Performance Across Parasite Taxa
| Parasite Group | Genera/Species Detected | Detection Efficiency | Notes/Applications |
|---|---|---|---|
| Apicomplexans | Plasmodium spp., Babesia spp. | High sensitivity for human-infecting species [10] | Detected mixed Plasmodium infections; identified Babesia in PCR-negative samples [3] [20] |
| Kinetoplastids | Trypanosoma cruzi, Leishmania spp. | Effectively detected in blood [10] | Identification to species level possible [10] |
| Filarial Nematodes | Various filarial species | Effectively detected in blood [10] | Microfilariae detection demonstrated [10] |
| Helminths | Various nematodes | Confirmed in mammalian samples [3] | Applied to wildlife surveillance [3] |
Detailed nUPDx Protocol
The nUPDx protocol involves a meticulously optimized sequence of enzymatic and amplification steps designed to maximize parasite DNA detection while minimizing host background interference.
Step-by-Step Experimental Procedure
Sample Preparation and Initial Digestion
- DNA Extraction: Extract total DNA from clinical specimens (200 μL blood recommended) using standardized extraction methods. Elute in 100 μL Tris/EDTA buffer [10].
- Primary Restriction Digestion (D1):
- Prepare digestion mixture containing:
- Total DNA extract (up to 40 μL)
- 5 μL PstI restriction enzyme buffer
- 1 μL PstI restriction enzyme (20 units)
- Nuclease-free water to 50 μL total volume
- Incubate at 37°C for 60 minutes
- Enzyme inactivation at 65°C for 20 minutes [10]
- Prepare digestion mixture containing:
Primary PCR Amplification
- First PCR with Outer Primers:
- Prepare reaction mixture:
- 5 μL digested DNA template
- 12.5 μL 2× PCR master mix
- 150 nM each outer pan-eukaryotic primer (without sequencing adapters)
- Nuclease-free water to 25 μL total volume
- Cycling conditions:
- Initial denaturation: 94°C for 3 minutes
- 25 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing: 52°C for 30 seconds
- Extension: 68°C for 60 seconds
- Final extension: 68°C for 5 minutes [10]
- Prepare reaction mixture:
Secondary Digestion and Nested PCR
Secondary Restriction Digestion (D2):
- Transfer 20 μL of first PCR product to new tube
- Add:
- 3 μL BsoBI restriction enzyme buffer
- 1 μL BsoBI restriction enzyme (10 units)
- 6 μL nuclease-free water
- Incubate at 37°C for 60 minutes
- Enzyme inactivation at 65°C for 20 minutes [10]
Second PCR with Inner Primers:
- Prepare reaction mixture:
- 5 μL secondary digested template
- 12.5 μL 2× PCR master mix
- 150 nM each inner primer (with full Illumina adapters and barcodes)
- Nuclease-free water to 25 μL total volume
- Cycling conditions:
- Initial denaturation: 94°C for 3 minutes
- 35 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing: 55°C for 30 seconds
- Extension: 68°C for 60 seconds
- Final extension: 68°C for 5 minutes [10]
- Prepare reaction mixture:
Library Preparation and Sequencing
Library Normalization and Pooling:
- Visualize PCR products by agarose gel electrophoresis
- Normalize amplicon concentrations using gel densitometry or fluorometric methods
- Pool normalized amplicons in equimolar ratios
Sequencing:
- Purify pooled library using DNA Clean and Concentrator Kit
- Quantify using Qubit Fluorometer
- Dilute to 4 nM and denature with 0.2 N NaOH
- Dilute to 20 pM and spike with 10% PhiX control DNA
- Sequence on Illumina MiSeq platform using 500-cycle v2 reagent kit [10]
Bioinformatic Analysis Pipeline
- Data Processing:
- Demultiplex sequences based on sample-specific barcodes
- Perform quality filtering and trimming
- Assemble paired-end reads
- Remove chimeric sequences using reference-based methods
- Classify sequences against curated 18S rDNA database (e.g., SILVA)
- Generate operational taxonomic units (OTUs) with 97% similarity threshold [10]
Essential Research Reagents and Materials
Table 3: Research Reagent Solutions for nUPDx Implementation
| Reagent Category | Specific Products | Function/Application | Notes |
|---|---|---|---|
| Restriction Enzymes | PstI, BsoBI | Selective host DNA digestion | BsoBI insensitive to CpG methylation [10] |
| PCR Master Mix | HotMasterMix (5Prime) | Robust amplification | Provides consistent performance across GC-rich templates [22] |
| Primer Sets | Custom pan-eukaryotic 18S rDNA primers | Universal parasite detection | Outer set without adapters; inner set with full Illumina adapters [10] |
| DNA Extraction | Qiagen EZ1 Advanced with DNA Tissue Kit | Total nucleic acid extraction | Optimized for blood specimens [22] |
| Library Purification | Zymo DNA Clean & Concentrator | PCR product clean-up | Efficient recovery of ~200-bp amplicons [10] |
| Quantification | Qubit Fluorometer 2.0 | Accurate DNA quantification | Preferred over spectrophotometry for low-concentration libraries [10] |
| Sequencing Platform | Illumina MiSeq | Amplicon sequencing | 500-cycle v2 reagent kit recommended [10] |
Applications and Implementation Considerations
The nUPDx assay has demonstrated utility across multiple research and diagnostic scenarios. In public health laboratory settings, the assay showed excellent concordance with real-time PCR methods when validating known positive specimens for Babesia microti, Trypanosoma cruzi, Leishmania tropica, and various Plasmodium species [20]. The method has been successfully applied to diverse biological specimens, including blood, tissues, and other sample types from mammals, birds, and reptiles [3], confirming its versatility for both clinical diagnostics and wildlife surveillance.
For optimal implementation, laboratories should consider the following:
- Sample Quality: Ensure adequate DNA input quantity and quality, particularly for processed specimens.
- Negative Controls: Include extraction and PCR-negative controls to monitor for contamination.
- Inhibition Assessment: Evaluate potential PCR inhibitors in complex matrices like whole blood.
- Database Curation: Maintain a comprehensive, curated database of parasite 18S rDNA sequences for accurate classification.
- Validation Framework: Establish laboratory-specific validation protocols using known positive controls.
The nUPDx technology represents a significant advancement in parasitic diagnostics, offering a universal detection approach with sensitivity comparable to pathogen-specific molecular methods. As next-generation sequencing becomes increasingly accessible and cost-effective, assays like nUPDx show great promise for comprehensive parasite detection in clinical, veterinary, and public health settings [20].
Universal parasite diagnostic (nUPDx) deep-amplicon sequencing represents a transformative approach in molecular parasitology, enabling the simultaneous detection and differentiation of diverse blood-borne parasites without prior knowledge of the specific infectious agent. This methodology addresses a critical diagnostic challenge: the genetic diversity of parasitic agents, which include protozoa and helminths, has historically complicated the development of broad-range detection assays. The nUPDx assay utilizes a sophisticated nested PCR technique targeting the 18S rDNA gene, coupled with restriction enzyme digestion to selectively deplete host-derived DNA, thereby enriching for parasite-derived sequences [23]. This application note details the experimental protocols and performance characteristics of nUPDx for detecting apicomplexans, kinetoplastids, and filarial nematodes, providing researchers with the necessary tools to implement this powerful diagnostic technology.
The fundamental innovation of the nUPDx assay lies in its combination of broad-range PCR amplification with strategic host-DNA depletion. The assay targets a ~200-bp region of the 18S rDNA gene, which contains restriction enzyme cut sites present in vertebrate hosts but absent in most blood parasites [23] [10]. This genetic distinction enables selective digestion of host 18S rDNA sequences prior to sequencing, significantly improving the relative abundance of parasite-derived reads.
The evolution of this technology has progressed through several key developments:
- Initial UPDx: A single-step PCR with one restriction digestion demonstrated feasibility but with sensitivity comparable to conventional PCR [10].
- Nested UPDx (nUPDx): Introduction of a nested PCR approach with two restriction digestion steps improved the limit of detection (LOD) approximately 10-fold, bringing it within the range of most qPCR methods [23].
- Adapter-incorporating UPDx (Ad_UPDx): Recent modifications integrate Illumina sequencing adapters during PCR amplification, reducing costs from approximately $40 to $11 per sample and decreasing turnaround time [16].
Table 1: Evolution of UPDx Assay Performance Characteristics
| Assay Version | Limit of Detection | Cost per Sample | Turnaround Time | Key Innovation |
|---|---|---|---|---|
| Initial UPDx | Comparable to conventional PCR | ~$40 | 7 days | Single PCR with one restriction digest |
| nUPDx | ~10-fold improvement over UPDx (0.58 parasites/μL for P. falciparum) | ~$40 | 7 days | Nested PCR with two restriction digests |
| Ad_UPDx | Similar to nUPDx (0.58 parasites/μL for P. falciparum) | ~$11 | 5 days | Integrated library preparation in PCR |
Experimental Protocols
Sample Preparation and DNA Extraction
Principle: High-quality DNA extraction is critical for successful nUPDx analysis. The protocol is optimized for whole blood specimens but has been successfully applied to various biological matrices including tissues and cultured parasites [3] [24].
Detailed Protocol:
- Sample Input: Use 200 μL of EDTA-preserved whole blood. For alternative specimens (tissues, cultured parasites), use approximately 25 mg of material.
- DNA Extraction: Perform extraction using the QIAamp DNA Mini Kit (Qiagen, Cat. #51306) on a QIAcube instrument.
- Critical Modification: Program the QIAcube for "no tip reuse" to eliminate potential cross-contamination between samples.
- Elution: Elute DNA in 100-200 μL of AE buffer. Store extracts at -20°C if not used immediately.
Validation: Include negative controls (parasite-free blood from healthy donors) and positive controls (blood spiked with known parasite cultures) in each extraction batch [16].
nUPDx Wet-Lab Procedure
Principle: The nUPDx assay employs a nested PCR approach with two sequential restriction enzyme digestions to deplete host DNA, thereby significantly enriching parasite-derived amplicons [23].
Reagents and Equipment:
- Restriction enzymes: PstI, BsoBI (New England Biolabs)
- PCR reagents: High-fidelity DNA polymerase, dNTPs, reaction buffer
- Primers: Custom pan-eukaryotic primers targeting 18S rDNA (sequences provided in Table 2)
- Thermal cycler
- Magnetic bead-based purification system
Detailed Protocol:
Step 1: Primary Restriction Digestion (D1)
- Prepare reaction mixture:
- Total DNA extract: 5 μL
- PstI restriction enzyme: 1 μL
- 10X CutSmart Buffer: 5 μL
- Nuclease-free water: to 50 μL total volume
- Incubate at 37°C for 60 minutes
- Purify using magnetic beads and elute in 20 μL nuclease-free water
Step 2: First PCR Amplification
- Prepare reaction mixture:
- Digested DNA: 5 μL
- Outer forward primer UPDx 18S Full F (10 μM): 1.25 μL
- Outer reverse primer UPDx 18S Full R (10 μM): 1.25 μL
- dNTPs (10 mM): 1 μL
- 5X HF Buffer: 10 μL
- High-fidelity DNA polymerase: 0.5 μL
- Nuclease-free water: to 50 μL total volume
- PCR conditions:
- Initial denaturation: 98°C for 30 seconds
- 25 cycles: 98°C for 10 seconds, 58°C for 20 seconds, 72°C for 20 seconds
- Final extension: 72°C for 5 minutes
- Purify PCR product using magnetic beads and elute in 20 μL nuclease-free water
Step 3: Secondary Restriction Digestion (D2)
- Prepare reaction mixture:
- First PCR product: 5 μL
- BsoBI restriction enzyme: 1 μL
- 10X Buffer: 5 μL
- Nuclease-free water: to 50 μL total volume
- Incubate at 37°C for 60 minutes
- Purify using magnetic beads and elute in 20 μL nuclease-free water
Step 4: Second PCR Amplification
- Prepare reaction mixture:
- Digested first PCR product: 5 μL
- Inner forward primer UPDx 18Sov F (10 μM): 1.25 μL
- Inner reverse primer UPDx 18Sov R (10 μM): 1.25 μL
- dNTPs (10 mM): 1 μL
- 5X HF Buffer: 10 μL
- High-fidelity DNA polymerase: 0.5 μL
- Nuclease-free water: to 50 μL total volume
- PCR conditions:
- Initial denaturation: 98°C for 30 seconds
- 35 cycles: 98°C for 10 seconds, 65°C for 20 seconds, 72°C for 20 seconds
- Final extension: 72°C for 5 minutes
- Purify final PCR product using magnetic beads and elute in 30 μL nuclease-free water
Sequencing and Bioinformatics Analysis
Principle: The nested PCR generates a ~200-bp amplicon suitable for deep sequencing on Illumina platforms. Bioinformatic analysis involves clustering sequences into operational taxonomic units (OTUs) and comparing them to reference databases for parasite identification [23] [16].
Library Preparation and Sequencing:
- For traditional nUPDx: Use Illumina library preparation kit following manufacturer's instructions
- For Ad_UPDx: Incorporate Illumina sequencing adapters during the second PCR using modified primers [16]
- Sequence on Illumina MiSeq platform with 2×250 bp paired-end reads
Bioinformatic Analysis Pipeline:
- Quality Control: Filter raw reads based on quality scores using Trimmomatic or similar tool
- Merge Paired-end Reads: Use PEAR or similar software
- Cluster OTUs: Use VSEARCH or USEARCH at 97% similarity threshold
- Taxonomic Assignment: BLAST comparison against curated 18S rDNA database
- Report Generation: Generate table of detected parasites and their relative read abundances
Research Reagent Solutions
Table 2: Essential Research Reagents for nUPDx Implementation
| Reagent/Equipment | Function | Specifications | Example Product |
|---|---|---|---|
| DNA Extraction Kit | Isolation of high-quality genomic DNA from clinical samples | Optimized for blood and tissues; silica-membrane technology | QIAamp DNA Mini Kit (Qiagen #51306) |
| Restriction Enzymes | Selective digestion of host 18S rDNA amplicons | High-fidelity; specific for vertebrate restriction sites | PstI, BsoBI (New England Biolabs) |
| Pan-Eukaryotic Primers | Amplification of 18S rDNA from diverse parasites | Target conserved regions flanking variable sequences; adapter-modified for Ad_UPDx | Custom sequences (see Table 3) |
| High-Fidelity DNA Polymerase | Accurate amplification of target region | Low error rate; robust performance with GC-rich templates | Phusion or Q5 Polymerase (NEB) |
| Magnetic Beads | Purification of DNA between enzymatic steps | Size-selective cleanup; high recovery efficiency | AMPure XP Beads (Beckman Coulter) |
| Sequencing Platform | Deep sequencing of amplicon libraries | Minimum 2×250 bp read length; high cluster density | Illumina MiSeq System |
Table 3: Primer Sequences for nUPDx Assay
| Primer Name | Sequence (5' to 3') | Application | Reference |
|---|---|---|---|
| UPDx 18S Full F | TTGATCCTGCCAGTAGTCATATGC | Outer forward primer (1st PCR) | [24] |
| UPDx 18S Full R | GGTGTGTACAAAGGGCAGGGAC | Outer reverse primer (1st PCR) | [24] |
| UPDx 18Sov F | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCGGAGAGGGAGCCTGAGA | Inner forward primer (2nd PCR) | [24] |
| UPDx 18Sov R | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGAGTCTCCGCTATCGGACG | Inner reverse primer (2nd PCR) | [24] |
Performance Characteristics
Analytical Sensitivity and Detection Limits
The nUPDx assay demonstrates exceptional sensitivity for diverse blood parasites, with a limit of detection (LOD) determined to be approximately 0.58 Plasmodium falciparum parasites/μL of blood, equivalent to 10-100 parasite genomes per reaction [23] [16]. This sensitivity falls within the range of most pathogen-specific qPCR assays, making it suitable for clinical applications.
Table 4: Detection Performance of nUPDx for Various Parasite Groups
| Parasite Group | Representative Species | Sample Type | Detection Concordance | Notes |
|---|---|---|---|---|
| Apicomplexans | Plasmodium falciparum, P. vivax, P. ovale, P. malariae, Babesia microti, B. divergens | Human blood | 100% for single infections; >85% for mixed infections | Species-level differentiation possible [16] [24] |
| Kinetoplastids | Trypanosoma cruzi, Leishmania tropica, Leishmania sp. | Human blood, cultured isolates | ~95% (1/13 T. cruzi samples missed) | Effective for acute and chronic infections [16] [24] |
| Filarial Nematodes | Loa loa, Brugia malayi | Human blood | 100% | Specific identification to species level [23] [16] |
| Mixed Infections | P. falciparum/P. ovale, P. falciparum/P. vivax, P. falciparum/P. malariae | Human blood | >85% | Occasionally detects only one species in mixed infections [16] [24] |
Applications Beyond Human Blood
The utility of nUPDx extends beyond human clinical diagnostics. Recent applications demonstrate its effectiveness in veterinary and wildlife contexts [3]:
- Mammalian samples: nUPDx confirmed apicomplexan and/or nematode infections in 24 of 32 parasite-positive mammals
- Avian and reptile samples: Detected infections in 6 of 13 positive bird and 1 of 2 positive reptile samples
- Whole parasites: Correctly identified 10 whole parasite specimens (worms and arthropods) to genus or family level, detecting one incorrect morphological identification
Technical Considerations and Limitations
While nUPDx represents a significant advancement in parasite detection, researchers should consider several technical aspects:
Host DNA Interference: Despite restriction digestion, host DNA may still constitute a substantial portion of sequences in high-host DNA samples (e.g., tissues). The nested approach with dual digestions typically reduces host-derived reads by >50% compared to undigested controls [23].
Primer Specificity: The pan-eukaryotic primers may not detect all parasite groups with equal efficiency due to primer-template mismatches. This limitation was observed for trichomonads and amoebae in cloacal swabs [3].
Sample Throughput: The traditional nUPDx protocol requires approximately 7 days from DNA extraction to results. The Ad_UPDx modification reduces this to 5 days while maintaining sensitivity [16].
Cost Considerations: Implementation requires significant initial investment in sequencing infrastructure. The Ad_UPDx modification reduces per-sample cost to approximately $11, making it more accessible for routine use [16].
Workflow Visualization
The nUPDx deep-amplicon sequencing technology represents a significant advancement in parasitology diagnostics, offering researchers a powerful tool for comprehensive detection of apicomplexans, kinetoplastids, and filarial nematodes. Its unique combination of broad-range PCR amplification with selective host-DNA depletion enables sensitive identification of single and mixed infections that might be missed by traditional methods. As sequencing costs continue to decrease and bioinformatic tools become more accessible, nUPDx and its derivatives hold promise for becoming standard tools in reference diagnostic laboratories, epidemiological surveillance programs, and research investigating parasite biodiversity. The protocols and performance data presented herein provide a foundation for researchers seeking to implement this technology in their investigative workflows.
Implementing nUPDx: From Sample to Sequence-Ready Library
The accuracy of deep-amplicon sequencing in universal parasite diagnostic (nUPDx) research is fundamentally dependent on the quality and purity of the input DNA. Effective sample preparation and DNA extraction are therefore critical first steps for successful pathogen identification, especially when working with diverse sample types such as blood, tissues, and dried blood spots (DBS) collected from a variety of animal hosts [6] [3]. The nUPDx approach, which relies on PCR amplification of the 18S rDNA gene followed by deep-amplicon sequencing, has demonstrated considerable promise for detecting parasitic infections in animals, identifying previously undetected apicomplexans and coinfections in mammals, birds, and reptiles [6]. However, its efficacy is highly contingent on the DNA extraction methodology employed, as different protocols can yield significantly different quantities and qualities of genomic material, directly impacting downstream diagnostic sensitivity [25] [26]. This application note provides detailed protocols and data-driven recommendations for optimizing DNA extraction from these key sample types within the context of nUPDx research.
Comparative Performance of DNA Extraction Methods
The selection of an appropriate DNA extraction method requires careful consideration of sample type, desired yield, and intended downstream application. The following tables summarize key performance data from published studies to guide this decision-making process.
Table 1: DNA Recovery Efficiency from Dried Blood Spots (DBS) using Different Extraction Kits
| Extraction Kit | Sample Type | Target DNA | Performance Metric (Cq Value Difference: WB vs. DBS) | Key Advantage |
|---|---|---|---|---|
| Qiagen DNeasy Blood & Tissue [25] | Canine WB (Spiked) | T. cruzi | Medium load: 0.25; High load: 0.65 | Most consistent recovery from filter paper |
| Zymo Quick-DNA/RNA Pathogen [25] | Canine WB (Spiked) | T. cruzi | Medium load: 1.10; High load: 5.22 | - |
| Chelex 100 Resin Method [27] | Human DBS | HIV-1 | Sensitivity: 90% vs. PBMC PCR | Rapid, cost-effective, no specialized equipment |
Table 2: Detection Sensitivity of nUPDx on Various Animal Specimens Following DNA Extraction
| Host Group | Microscopy/PCR-Positive Samples | nUPDx Confirmed Infections | Additional Coinfections Detected | Notes |
|---|---|---|---|---|
| Mammals [6] | 32 | 24 | Several | Successful detection of apicomplexan and nematode infections. |
| Birds [6] | 13 | 6 | - | - |
| Reptiles [6] | 2 | 1 | - | - |
| Whole Parasites [6] | 10 (by morphology) | 10 to genus/family level | 1 incorrect morphology ID corrected | Demonstrates utility for specimen identification. |
Detailed Experimental Protocols
Protocol A: Rapid DNA Extraction from Dried Blood Spots using Chelex Resin
This protocol, adapted from published methods for HIV-1 diagnosis [27] and parasite research [28], provides a simple and cost-effective means of DNA extraction.
Principle: Chelex resin acts as a chelating agent that binds metal ions, which are co-factors for DNases, thereby protecting DNA from degradation. The boiling step lyses cells and denatures proteins, releasing DNA into solution [28].
Materials:
- Punched DBS (3-6 mm) from filter paper (e.g., Whatman FTA, Isocode, or Nobuto strip)
- Chelex 100 Resin (5-6.7% suspension in DNase-free water) [27] [28]
- 0.5% Saponin in DNase-free water
- Phosphate Buffered Saline (PBS)
- Microcentrifuge tubes (1.5 mL and 0.6 mL)
- Thermo-shaker or heat block
- Microcentrifuge
- Aerosol-barrier pipette tips
Procedure:
- Erythrocyte Lysis: Place one punched DBS into a 1.5 mL microcentrifuge tube. Add 1 mL of 0.5% saponin. Vortex and incubate at 4°C for a minimum of 4 hours or overnight [28].
- Washing: Centrifuge the tube briefly (e.g., 10 seconds at 4000 rpm). Carefully aspirate and discard the saponin supernatant. Add 1 mL of PBS to the tube, vortex, and incubate at 4°C for 20-30 minutes. Centrifuge briefly and aspirate the PBS supernatant, leaving the filter paper disk in the tube [28].
- DNA Elution: Add 150-200 µL of 5% Chelex resin suspension to the tube [27] [28].
- Incubation: Incubate the tube in a thermo-shaker at 95-100°C for 10-30 minutes [27] [28]. Important: If using a sealed heat block, open the tube lid twice during the first few minutes to release steam and prevent pressure buildup.
- Bead Separation: Centrifuge the tube at 4000 rpm for 5 minutes to pellet the Chelex beads.
- Supernatant Transfer: Carefully transfer the supernatant (containing the DNA) to a new 0.6 mL tube using an aerosol-barrier tip, avoiding the transfer of any beads.
- Final Clearance: Centrifuge the 0.6 mL tube again at 4000 rpm for 10 minutes. Transfer the final cleared supernatant (approx. 100 µL) to a fresh, labeled microcentrifuge tube.
- Storage: Store extracted DNA at -20°C for immediate use or -80°C for long-term storage.
Protocol B: Optimized Column-Based DNA Extraction from Dried Blood Spots
For applications requiring high-purity DNA, such as deep-amplicon sequencing, column-based purification is often preferred. This protocol is optimized for Nobuto filter strips and the Qiagen DNeasy Blood & Tissue Kit [25].
Materials:
- Qiagen DNeasy Blood & Tissue Kit
- Punched DBS from Nobuto filter strip
- Proteinase K
- Ethanol (96-100%)
- Water bath or thermo-shaker
Procedure:
- Initial Incubation: Place the punched DBS in a 1.5 mL microcentrifuge tube. Add 180 µL of Buffer ATL from the kit. Incubate at 90°C for 10 minutes without agitation [25].
- Lysis: Add 25 µL of Proteinase K to the tube. Mix by vortexing thoroughly.
- Extended Digestion: Incubate at 56°C for an extended period (e.g., 3 hours to overnight) with constant shaking [25].
- Continue per Manufacturer's Instructions: After digestion, add 200 µL of Buffer AL and 200 µL of ethanol. Mix thoroughly and transfer the entire mixture to a DNeasy Mini spin column.
- Washes and Elution: Continue with the standard kit protocol, including wash steps with Buffers AW1 and AW2. Elute DNA in a small volume (e.g., 50-100 µL) of Buffer AE or DNase-free water.
nUPDx Application on Animal Specimens
The universal parasite diagnostic test (nUPDx) can be applied to DNA extracted from a wide range of biological specimens from animals [6] [3].
Workflow:
- Sample Collection: Collect blood (on filter paper or as liquid blood), tissues, or other biological samples from the target animal.
- DNA Extraction: Use an appropriate extraction method (e.g., Protocol A or B) to obtain total genomic DNA. For tissue samples, a standard column-based kit with an extended proteinase K digestion is recommended.
- PCR Amplification: Perform PCR amplification of the 18S rDNA gene using the universal primers specified in the nUPDx assay.
- Deep-Amplicon Sequencing: Prepare libraries from the PCR products and sequence on an Illumina platform.
- Bioinformatic Analysis: Process the sequencing data through the nUPDx bioinformatic pipeline to assign reads to specific parasite genera or species.
Note: The nUPDx assay may not detect all parasites due to primer-template mismatches, as was observed with trichomonads and amoebae in bird and reptile samples [6]. It is therefore recommended to use nUPDx in conjunction with other diagnostic methods for comprehensive parasite screening.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for DNA Extraction in Parasite Diagnostics
| Reagent / Material | Function | Application Example |
|---|---|---|
| Chelex 100 Resin [27] [28] | Chelates metal ions to inhibit DNases; simplifies DNA release via boiling. | Rapid DNA extraction from DBS for PCR-based screening. |
| Nobuto Blood Filter Strips [25] | Cost-effective filter paper for stable room-temperature storage of whole blood. | Field collection and biobanking of blood from wildlife reservoirs. |
| Saponin [28] | Lyses erythrocytes by complexing with membrane cholesterol. | Releasing intra-erythrocytic parasites (e.g., Babesia, malaria) during DBS processing. |
| Proteinase K [25] | Digest proteins and nucleases for comprehensive tissue lysis. | Efficient digestion of animal tissues and blood clots in optimized kit protocols. |
| Qiagen DNeasy Blood & Tissue Kit [25] | Silica-membrane column for purifying high-quality, PCR-ready DNA. | Optimal recovery of parasite and host DNA for sensitive nUPDx sequencing. |
Workflow Visualization
DNA Extraction Workflow for nUPDx
nUPDx Analysis Post-Extraction
Nested Polymerase Chain Reaction (PCR) is a highly sensitive and specific molecular technique that significantly reduces non-specific amplification by using two sequential rounds of PCR amplification with two sets of primers [29]. This method is particularly valuable in diagnostic applications where target DNA may be present in minimal quantities or amid significant background DNA, such as in the identification of parasitic pathogens [30] [5]. Within the context of universal parasite diagnostic (nUPDx) deep-amplicon sequencing research, nested PCR serves as a critical enrichment step, enabling the selective amplification of parasite-derived 18S rDNA targets from complex biological samples [5] [3]. The incorporation of restriction digestion steps further enhances assay specificity by selectively depleting abundant host DNA, thereby increasing the proportion of parasite sequences available for downstream sequencing [5]. This application note provides a detailed protocol for implementing nested PCR within nUPDx workflows, specifically focusing on primer design strategies, optimized cycling conditions, and integrated restriction enzyme digestion steps to support reliable parasite detection and identification.
Experimental Principles and Workflow
Fundamental Principles of Nested PCR
The enhanced specificity of nested PCR stems from its two-stage amplification approach [29]. The initial round of PCR utilizes an outer primer pair that flanks the target region, generating an intermediate amplicon that contains the specific target sequence along with potential non-specific products [31]. A small aliquot of this first reaction is then transferred to a second PCR containing inner primers that bind within the initial amplicon [30]. This sequential priming strategy dramatically reduces false-positive results because it is statistically improbable that non-specific amplification products from the first reaction would contain binding sites for the second set of primers [31]. In nUPDx applications, this principle is leveraged to amplify parasite DNA from clinical specimens where host DNA predominates, thereby improving the detection limit for pathogenic organisms [5] [24].
Integration with Restriction Digestion in nUPDx
In the specialized context of nUPDx research, nested PCR is coupled with restriction enzyme digestion to create a powerful selective enrichment strategy [5]. The eukaryotic 18S rDNA gene, while conserved across species, contains single nucleotide polymorphisms that differentiate host from parasite sequences [24]. Following the initial PCR amplification with universal eukaryotic primers, restriction enzymes are employed that specifically target and cleave host-derived 18S rDNA amplicons at these polymorphic sites [5]. This digestion renders host DNA unsuitable for subsequent amplification while leaving parasite DNA intact. The nested PCR then selectively amplifies the undigested parasite templates, significantly enriching parasite signal and improving the sensitivity of detection in downstream sequencing applications [5] [24].
Visual Workflow Representation
The following diagram illustrates the complete integrated workflow of nested PCR with restriction digestion within the nUPDx paradigm:
Figure 1: Integrated nUPDx workflow combining nested PCR with restriction digestion for parasite detection.
Materials and Reagents
Research Reagent Solutions
The following table details essential reagents required for implementing the nested PCR workflow with restriction digestion:
Table 1: Essential research reagents for nested PCR in nUPDx applications
| Reagent Category | Specific Examples | Function in Workflow |
|---|---|---|
| Polymerase Enzymes | Hot-start DNA polymerase (e.g., Platinum II Taq) [29] | Reduces non-specific amplification during reaction setup; essential for complex multiplex reactions |
| Restriction Enzymes | Type II restriction enzymes [32] | Selective digestion of host 18S rDNA amplicons based on sequence polymorphisms [5] |
| Primer Sets | Outer primers: Universal eukaryotic 18S rDNA [24]Inner primers: Parasite-specific 18S rDNA [5] | Target amplification with increasing specificity through sequential rounds |
| Buffer Systems | Isothermal Amplification Buffer [33]Restriction Enzyme Buffers [32] | Maintain optimal enzyme activity and fidelity during amplification and digestion steps |
| Nucleotides & Cofactors | dNTPs, MgCl₂, MgSO₄ [33] | Essential components for DNA synthesis and polymerase activity |
| DNA Extraction Kits | QIAamp DNA Mini Kit [24] | High-quality DNA extraction from clinical specimens (blood, tissues) |
| Purification Kits | Gel extraction, PCR clean-up kits | Purification of amplification products between procedural steps |
Detailed Methodologies
Primer Design and Optimization
Effective primer design is fundamental to successful nested PCR applications in parasite diagnostics. The following specifications ensure optimal performance:
Outer Primer Design: Target universal regions of the 18S small subunit ribosomal RNA (SSU rRNA) gene with sequences such as 5'-TTGATCCTGCCAGTAGTCATATGC-3' (forward) and 5'-GGTGTGTACAAAGGGCAGGGAC-3' (reverse) to broadly amplify eukaryotic DNA [24]. These should generate amplicons of 300-500 bp for optimal downstream processing [30].
Inner Primer Design: Design nested primers to bind approximately 50-100 bp internal to the outer primer binding sites [31]. For parasite-specific detection, target genetic markers such as the CYP51C gene for Fusarium tricinctum or variable regions of the 18S rDNA for Plasmodium species differentiation [30] [34].
Primer Validation: Conduct rigorous in silico validation using Primer-BLAST against relevant genomic databases to ensure specificity for target parasites [30]. Empirically verify primer performance through temperature gradient PCR to establish optimal annealing conditions before implementing in diagnostic workflows.
PCR Cycling Conditions and Parameters
The following table provides detailed cycling parameters for both rounds of nested PCR amplification:
Table 2: Optimized cycling conditions for nested PCR in parasite detection
| Parameter | First PCR Round (Outer Primers) | Second PCR Round (Nested Primers) |
|---|---|---|
| Initial Denaturation | 95°C for 5 min [34] | 95°C for 4 min [34] |
| Cycle Count | 40 cycles [34] | 35 cycles [34] |
| Denaturation | 94°C for 45 s [34] | 94°C for 20 s [34] |
| Annealing | 60°C for 45 s [34] | 60°C for 20 s [34] |
| Extension | 72°C for 70 s [34] | 72°C for 45 s [34] |
| Final Extension | 72°C for 10 min [34] | 72°C for 10 min [34] |
| Template Volume | 10 ng genomic DNA [34] | 1:1000 dilution of first PCR product [34] |
Restriction Digestion Integration
The restriction digestion step is strategically implemented between the two PCR rounds to selectively deplete host-derived amplicons:
Enzyme Selection: Choose restriction enzymes that recognize and cleave specific sequence polymorphisms present in host 18S rDNA but absent in target parasite sequences [5]. This selective digestion enriches the relative abundance of parasite templates.
Reaction Setup: Following the first PCR round, transfer 5-10 μL of amplification product to a fresh tube containing 1-2 units of selected restriction enzyme(s) and appropriate reaction buffer [5]. Incubate at the recommended temperature (typically 37°C) for 30-60 minutes.
Process Integration: Following restriction digestion, the reaction mixture can be used directly as template for the second PCR round without purification [5]. The nested primers will preferentially amplify intact (parasite-derived) templates, while digested host amplicons will not support amplification.
Technical Considerations
Contamination Prevention
- Physical Separation: Perform reagent preparation, first PCR, restriction digestion, and second PCR in physically separated work areas with dedicated equipment [30].
- Negative Controls: Include multiple negative controls (no-template and no-enzyme) across all procedural steps to monitor for contamination [5].
- Aerosol Prevention: Use barrier tips and closed-tube systems during liquid transfers to prevent amplicon carryover between reactions [24].
Sensitivity and Specificity Optimization
- Cycle Number Balance: Limit cycle numbers in both amplification rounds (typically 30-40 cycles for round one, 25-35 cycles for round two) to minimize non-specific amplification while maintaining detection sensitivity [34].
- Hot-Start Implementation: Utilize hot-start DNA polymerases to prevent primer-dimer formation and mispriming during reaction setup, particularly important for complex clinical samples [29].
- Template Dilution: Employ appropriate dilution of first-round products (typically 1:100 to 1:1000) when transferring to the second reaction to prevent polymerase inhibition and maintain reaction efficiency [34].
Applications in Parasite Diagnostics
The integrated nested PCR with restriction digestion workflow has demonstrated particular utility in universal parasite detection applications:
- Multi-Parasite Detection: The nUPDx approach has successfully identified diverse blood parasites including Plasmodium spp., Babesia spp., kinetoplastids, and filarial nematodes from clinical specimens [5] [24].
- Sensitive Detection: This methodology achieves detection limits as low as 0.58 Plasmodium falciparum parasites/μL of blood, surpassing the sensitivity of conventional microscopy and comparable to real-time PCR assays [5].
- Mixed Infection Resolution: The deep-amplicon sequencing following nested PCR enrichment enables detection and differentiation of mixed parasite infections that may be missed by species-specific PCR assays [3] [24].
- Host DNA Depletion: The restriction digestion step typically reduces host-derived reads by 80-95% in sequencing outputs, dramatically enriching for parasite sequences and improving diagnostic confidence [5].
Universal Parasite Diagnostic (UPDx) deep-amplicon sequencing represents a significant advancement in the detection and differentiation of parasitic pathogens from complex clinical samples. The foundational UPDx assay utilized a targeted amplicon deep sequencing (TADS) strategy, employing pan-eukaryotic primers to amplify the 18S rDNA gene segment, followed by restriction enzyme digestion to selectively reduce host-derived DNA [24]. While this approach demonstrated promise for detecting diverse blood parasites including Plasmodium spp., Babesia spp., kinetoplastids, and filarial nematodes, its routine application in diagnostic laboratories faced challenges due to lengthy procedures and considerable expenses [5]. The evolution of this methodology into a nested PCR approach (nUPDx) improved sensitivity by approximately tenfold, achieving a limit of detection (LOD) comparable to most real-time PCR methods [10].
The AdUPDx protocol represents a further refinement of this diagnostic approach, specifically designed to enhance efficiency and cost-effectiveness. This method incorporates Illumina barcodes and adapters directly during the PCR amplification steps, creating amplicons that are immediately ready for sequencing on Illumina platforms [5]. This strategic integration eliminates the requirement for a separate, dedicated library preparation step—a process that is both time-consuming and financially burdensome. By streamlining the workflow, AdUPDx substantially reduces both the turnaround time and operational costs associated with parasitic diagnostics, making it more accessible for routine laboratory use. The development of Ad_UPDx is particularly valuable for public health and reference laboratories that receive specimens from patients with potential exposure to diverse parasitic pathogens, where comprehensive testing capabilities are essential [24].
Key Research Reagent Solutions
The successful implementation of Ad_UPDx relies on several critical reagents and components that facilitate the integrated library preparation and amplification process.
Table 1: Essential Research Reagents for Ad_UPDx Implementation
| Reagent Component | Function in Ad_UPDx Protocol |
|---|---|
| Modified Primer Sets | Primer pairs are redesigned to include Illumina adapter sequences, enabling direct incorporation during amplification [5]. |
| Illumina Barcodes/Indexes | Unique nucleotide sequences added to each sample, enabling multiplex sequencing and subsequent sample identification [35]. |
| Restriction Enzymes | Enzymes selectively digest host 18S rDNA based on vertebrate-specific cut sites, enriching parasite-derived amplicons [24] [10]. |
| Pan-Eukaryotic Primers | Target conserved regions of the 18S rDNA gene, enabling amplification of DNA from diverse parasite taxa [24]. |
| Nested PCR Reagents | Outer and inner primer sets enable two amplification rounds, improving sensitivity and providing opportunity for host DNA depletion [10]. |
Ad_UPDx Experimental Workflow and Protocol
The Ad_UPDx protocol integrates library preparation into the amplification process through a carefully designed sequence of enzymatic and amplification steps. The following workflow diagram outlines the key procedural stages:
Detailed Procedural Steps
DNA Extraction and Primary PCR: The process begins with the extraction of total DNA from patient specimens, typically from EDTA-preserved whole blood using commercial kits such as the QIAamp DNA Mini Kit [24]. The initial amplification employs outer pan-eukaryotic primers (e.g., UPDx 18S Full F: TTGATCCTGCCAGTAGTCATATGC and UPDx 18S Full R: GGTGTGTACAAAGGGCAGGGAC) that target a broad region of the 18S rDNA gene [24]. This first PCR amplifies DNA from both host and potential parasites present in the sample.
Restriction Digestion of Host DNA: Following the primary PCR, a restriction enzyme digestion is performed using enzymes that target cut sites present specifically in vertebrate 18S rDNA sequences. The Ad_UPDx protocol utilizes PstI for the first digestion (D1), which acts on the larger amplicon generated by the outer primers [10]. This strategic step selectively cleaves host-derived amplicons, significantly reducing the background of human DNA that could otherwise dominate subsequent sequencing steps.
Nested PCR with Adapter-Integrated Primers: The critical innovation of Ad_UPDx occurs in the nested PCR step, which employs inner primers pre-modified with Illumina adapter sequences and barcodes. These primers (e.g., UPDx 16Sov F: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCGGAGAGGGAGCCTGAGA...) incorporate the necessary flow cell binding sites and sample-specific indices directly during amplification [5] [24]. This integration transforms the target amplicons into sequencing-ready libraries without requiring separate adapter ligation.
Secondary Restriction Digestion and Library Preparation: A second restriction digestion (D2) is performed using enzymes such as BsoBI or BamHI-HF, which target different vertebrate-specific cut sites within the nested amplicon [10]. This further depletes any residual host DNA, enriching the final library for parasite-derived sequences. The resulting digested amplicons are then quantified, normalized, and pooled for multiplexed sequencing on the Illumina MiSeq platform [5].
Performance Metrics and Validation Data
The Ad_UPDx methodology has been rigorously evaluated against previous iterations of the universal parasite diagnostic assay, demonstrating significant improvements in key performance indicators.
Table 2: Comparative Performance of Ad_UPDx Versus Previous Assay Versions
| Performance Parameter | Original TADS Assay [5] [10] | Ad_UPDx Modified Assay [5] |
|---|---|---|
| Total Turnaround Time | 7 days | 5 days |
| Cost Per Sample | ~$40 USD | ~$11 USD |
| Limit of Detection (LoD) | Comparable to conventional PCR | 0.58 P. falciparum/μL blood |
| Host DNA Depletion | Single restriction digestion | Dual restriction digestion strategy |
| Library Prep Requirement | Separate library preparation needed | Integrated into PCR amplification |
Diagnostic Accuracy and Applications
Validation studies have demonstrated that Ad_UPDx maintains excellent concordance with established diagnostic methods while expanding detection capabilities. In evaluations conducted at the Wadsworth Center Parasitology Laboratory, the assay correctly identified Babesia microti, Trypanosoma cruzi, Leishmania tropica, and various Plasmodium species in clinical specimens previously confirmed by real-time PCR [24]. The assay has also been successfully adapted for use with non-human samples, detecting apicomplexan and nematode infections in mammals, birds, and reptiles, thereby demonstrating its utility for veterinary diagnostics and wildlife surveillance [3].
The integration of Illumina barcodes enables high-level multiplexing, allowing numerous samples to be processed simultaneously in a single sequencing run. This sample multiplexing strategy dramatically increases throughput while reducing per-sample costs, making the assay particularly suitable for laboratories processing large sample volumes [35]. The bioinformatic analysis pipeline following sequencing utilizes the barcode information to demultiplex samples automatically, streamlining the path from raw sequence data to pathogen identification.
Discussion and Future Applications
The Ad_UPDx protocol represents a substantial innovation in parasitic diagnostic sequencing, effectively addressing two major constraints previously associated with next-generation sequencing approaches: cost and complexity. By eliminating the separate library preparation step, the assay achieves a remarkable 72.5% reduction in per-sample costs (from $40 to $11) and shortens turnaround time by approximately 40% (from 7 to 5 days) [5]. These improvements make deep-amplicon sequencing increasingly accessible for routine diagnostic applications, particularly in public health laboratories where comprehensive pathogen detection is essential.
Future developments of the AdUPDx methodology will likely focus on expanding its application beyond blood specimens. Preliminary research has already demonstrated its utility for detecting parasites in various tissue types [3], suggesting potential for adaptation to other clinical matrices such as feces and cerebrospinal fluid. Additionally, the core principle of integrating adapter sequences during amplification could be applied to other multiplex PCR-based diagnostic assays, potentially revolutionizing the efficiency of metagenomic sequencing for bacterial, viral, and fungal pathogens. As sequencing costs continue to decline and bioinformatic tools become more user-friendly, AdUPDx and similar integrated approaches are poised to become increasingly central to modern diagnostic parasitology.
The Illumina MiSeq System is a benchtop next-generation sequencing (NGS) platform that combines cluster generation, sequencing-by-synthesis chemistry, and data analysis in a single, compact instrument [36]. Its rapid turnaround time and operational simplicity make it particularly suitable for diagnostic applications, including the detection and differentiation of blood parasites through deep-amplicon sequencing approaches. For researchers focused on universal parasite diagnostic (nUPDx) research, the MiSeq offers a balance of read length, output, and ease of use that facilitates the identification of diverse parasitic organisms from clinical samples.
The utility of the MiSeq platform in parasitology has been demonstrated through its application in the nested Universal Parasite Diagnostic (nUPDx) assay, a targeted amplicon deep sequencing (TADS) strategy that enables simultaneous detection of apicomplexan parasites (Plasmodium spp., Babesia spp.), kinetoplastids, and filarial nematodes from blood specimens [16] [10]. This assay leverages the MiSeq's capabilities to provide species-level differentiation critical for proper patient management and treatment decisions. The system's flexibility in read length and flow cell configurations allows researchers to optimize sequencing runs specifically for parasite detection, maximizing efficiency while controlling costs.
MiSeq System Technical Specifications
Understanding the technical capabilities of the MiSeq system is essential for designing effective parasite detection assays. The platform offers multiple reagent kits with varying output capacities and run characteristics, allowing researchers to select parameters based on their specific project needs [37].
Output Specifications by Reagent Kit
Table 1: MiSeq System output specifications across different reagent kits
| Read Length | MiSeq Nano Kit v2 | MiSeq Micro Kit v2 | MiSeq Kit v2 | MiSeq Kit v3 |
|---|---|---|---|---|
| 2 × 150 bp | 300 Mb | 1.2 Gb | 4.5–5.1 Gb | N/A |
| 2 × 250 bp | 500 Mb | N/A | 7.5–8.5 Gb | N/A |
| 2 × 300 bp | N/A | N/A | N/A | 13.2–15 Gb |
Performance Metrics and Quality Scores
Table 2: MiSeq System performance characteristics and quality metrics
| Parameter | MiSeq Reagent Kit v2 | MiSeq Reagent Kit v3 |
|---|---|---|
| Maximum Reads | 24–30M (paired-end) | 44–50M (paired-end) |
| Run Time (2 × 250 bp) | ~39 hours | N/A |
| Run Time (2 × 300 bp) | N/A | ~56 hours |
| Quality Scores (2 × 250 bp) | >75% bases >Q30 | N/A |
| Quality Scores (2 × 300 bp) | N/A | >70% bases >Q30 |
The MiSeq system's 2 × 300 bp paired-end read capability using the v3 reagent kit is particularly advantageous for parasite detection assays, as it provides sufficient read length to cover the ~200-bp 18S rDNA target region with overlap, enabling high-confidence taxonomic assignment [36] [37]. The system's output of 13.2–15 Gb and 44–50 million paired-end reads per run using the v3 kit provides ample capacity for multiplexing numerous clinical samples in a single sequencing run [37].
Universal Parasite Diagnostic (nUPDx) Workflow
The nUPDx assay employs a nested PCR approach targeting the 18S rDNA gene, followed by high-throughput sequencing on the MiSeq platform. This method incorporates strategic restriction enzyme digestions that selectively reduce host-derived DNA, thereby enriching for parasite sequences and significantly enhancing detection sensitivity [10].
nUPDx Workflow Diagram
Key Workflow Steps
Initial Restriction Digestion: Genomic DNA extracts undergo first digestion with PstI restriction enzyme which targets vertebrate 18S rDNA sequences, reducing amplifiable host DNA before the first PCR [10].
First PCR Amplification: Using pan-eukaryotic outer primers (UPDx 18S Full F: TTGATCCTGCCAGTAGTCATATGC; UPDx 18S Full R: GGTGTGTACAAAGGGCAGGGAC) to amplify a larger ribosomal DNA region encompassing the target ~200-bp region [24].
Second Restriction Digestion: The first PCR product undergoes digestion with BsoBI restriction enzyme (replacing XmaI in original protocols), which further reduces host-derived amplicons by targeting additional vertebrate-specific restriction sites [10].
Second Nested PCR: Using inner primers that incorporate Illumina adapter sequences, enabling direct preparation of sequencing-ready libraries without separate library preparation steps. This step also incorporates unique dual indices for sample multiplexing [16].
Sequencing on MiSeq: Pooled libraries are sequenced using 2 × 250 bp or 2 × 300 bp paired-end chemistry on the MiSeq platform, providing sufficient read length and quality for accurate parasite identification [16] [37].
Adapter-Incorporating UPDx (Ad_UPDx) Optimization
Recent modifications to the nUPDx protocol have resulted in the development of adapter-incorporating UPDx (Ad_UPDx), which streamlines the workflow and reduces costs while maintaining sensitivity [16]. This optimized approach incorporates Illumina sequencing adapters directly during PCR amplification, eliminating the need for separate library preparation.
Ad_UPDx Experimental Protocol
Primer Design and Modification
Initial PCR Primers: Design outer primers with overhang adapter sequences (5'-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-[locus-specific sequence]-3' for forward primer; 5'-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-[locus-specific sequence]-3' for reverse primer) [16].
Nested PCR Primers: Use primers that incorporate full adapter sequences and unique dual indices for multiplexing, compatible with Illumina MiSeq sequencing: Forward Primer: 5'-AATGATACGGCGACCACCGAGATCTACAC[i5]TCGTCGGCAGCGTC-3' Reverse Primer: 5'-CAAGCAGAAGACGGCATACGAGAT[i7]GTCTCGTGGGCTCGG-3' where [i5] and [i7] represent unique 8-base indices [16].
PCR Amplification Conditions
First PCR Reaction:
- Reaction Volume: 25 μL
- Template DNA: 5 μL (≤100 ng)
- Primer Concentration: 0.5 μM each
- Cycling Conditions: 95°C for 3 min; 35 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s; final extension 72°C for 5 min [16]
Restriction Digestion Between PCRs:
- Enzyme: BsoBI (or BamHI-HF)
- Conditions: 37°C for 60 min, followed by enzyme inactivation at 65°C for 20 min [10]
Second PCR Reaction:
- Template: 2 μL of first PCR product (after restriction digestion)
- Primer Concentration: 0.5 μM each (indexing primers with full adapters)
- Cycling Conditions: 95°C for 3 min; 15 cycles of 95°C for 30 s, 60°C for 30 s, 72°C for 30 s; final extension 72°C for 5 min [16]
Library Preparation and Sequencing
- Library Normalization: Clean amplified products using AMPure XP beads, quantify by fluorometry, and normalize to 4 nM [16]
- Pooling and Denaturation: Combine equal volumes of normalized libraries, denature with 0.2 N NaOH, and dilute to optimal loading concentration (typically 8-12 pM for standard MiSeq, ~60 pM for MiSeq i100 when migrating from v3 chemistry) [16] [38]
- Sequencing Parameters: Use MiSeq Reagent Kit v3 (2 × 300 bp) with 15% PhiX spike-in to address low diversity amplicon libraries [16] [37]
Ad_UPDx Workflow Diagram
Performance Evaluation and Optimization Guidelines
Sensitivity and Specificity Assessment
The nUPDx and Ad_UPDx assays have been rigorously validated against conventional diagnostic methods including microscopy and pathogen-specific PCR assays [16] [24]. Performance metrics demonstrate their utility for clinical parasitology diagnostics:
Limit of Detection (LOD): The Ad_UPDx assay demonstrates a LOD of 0.58 Plasmodium falciparum parasites/μL of blood, comparable to real-time PCR methods and suitable for clinical diagnostics [16]
Species Differentiation: The assay successfully differentiates human-infecting Plasmodium species (P. falciparum, P. vivax, P. ovale, P. malariae), Babesia species (B. microti, B. divergens, B. duncani), kinetoplastids (Leishmania spp., Trypanosoma cruzi, Trypanosoma brucei), and filarial nematodes (Loa loa, Brugia malayi) [16] [10]
Concordance with Reference Methods: In validation studies comparing 36 clinical samples, Ad_UPDx showed complete concordance with reference methods for 33 samples (91.7%), with discordant results primarily in mixed-species infections where one species was detected but not both [16]
Cost and Time Efficiency Analysis
Table 3: Comparison of nUPDx and Ad_UPDx workflows for parasite detection
| Parameter | Original nUPDx | Optimized Ad_UPDx |
|---|---|---|
| Total Cost per Sample | ~$40 | ~$11 |
| Library Preparation Time | 3 days | 1 day |
| Total Turnaround Time | 7 days | 5 days |
| Laboratory Hands-on Time | 6–8 hours | 3–4 hours |
| Number of PCR Steps | 2 + separate library prep | 2 (incorporates library prep) |
| Compatibility with MiSeq | Requires separate library preparation | Direct loading after PCR |
The Ad_UPDx protocol reduces costs by approximately 72.5% (from $40 to $11 per sample) and decreases turnaround time by 2 days through the elimination of separate library preparation steps [16]. This significant improvement in efficiency makes deep-amplicon sequencing for parasite detection more accessible for routine diagnostic applications.
Loading Concentration Optimization
Optimal loading concentrations are critical for achieving high-quality sequencing data on the MiSeq platform. When using custom libraries like Ad_UPDx, titration experiments may be necessary:
- Standard Libraries: Typically load at 80-120 pM for conventional MiSeq systems [38]
- Migrating from MiSeq v3: Use multiplier of ~6.5X when transitioning to MiSeq i100 series [38]
- Custom Library Titration: For libraries not listed in validated protocols, perform titration experiments starting at calculated concentrations and adjust in 20 pM increments based on initial run performance [38]
Essential Research Reagent Solutions
Table 4: Key reagents and materials for nUPDx and Ad_UPDx assays
| Reagent/Material | Function | Specifications/Alternatives |
|---|---|---|
| QIAamp DNA Mini Kit | DNA extraction from whole blood | 200 μL input, 200 μL elution [24] |
| PstI Restriction Enzyme | First host DNA digestion | Targets vertebrate-specific 18S rDNA sites [10] |
| BsoBI Restriction Enzyme | Second host DNA digestion | Replaces XmaI; not sensitive to CpG methylation [10] |
| Pan-Eukaryotic Primers | 18S rDNA amplification | Outer: UPDx 18S Full F/R; Inner: Adapter-incorporated [24] |
| Illumina Index Primers | Sample multiplexing | Unique dual 8-base indices for sample identification [16] |
| AMPure XP Beads | PCR purification | Size selection and cleanup between steps [16] |
| MiSeq Reagent Kit v3 | Sequencing | 2 × 300 bp, 600-cycle (15 Gb output) [37] |
| PhiX Control Library | Sequencing control | 15% spike-in for low diversity libraries [37] |
The Illumina MiSeq platform provides an ideal balance of read length, output capacity, and operational simplicity for universal parasite detection using deep-amplicon sequencing approaches. The optimized Ad_UPDx protocol significantly reduces cost and turnaround time while maintaining sensitivity comparable to real-time PCR assays, making it increasingly amenable to routine diagnostic applications. As NGS technologies continue to advance and costs decrease, targeted amplicon sequencing approaches for comprehensive parasite detection are poised to become valuable tools in clinical diagnostics, reference laboratories, and public health surveillance systems.
The nested Universal Parasite Diagnostic (nUPDx) deep-amplicon sequencing represents a significant advancement in parasitology, enabling the detection and identification of diverse parasitic organisms from a single assay. Originally developed for human blood parasites, this method employs a nested PCR approach targeting the 18S rDNA gene, followed by deep sequencing to achieve a limit of detection within the range of most qPCR methods [10]. The core innovation lies in its use of restriction enzyme digestion to selectively deplete host DNA, thereby enhancing the amplification and detection of parasite-derived DNA [10]. This application note details the translation and validation of the nUPDx assay for use in veterinary diagnostics and wildlife surveillance, highlighting its utility in detecting unknown or unexpected pathogens that species-specific PCRs might miss [6] [3].
Application Notes: nUPDx in Animal Species
The nUPDx assay has been successfully applied to a variety of biological specimens from mammals, birds, and reptiles, demonstrating its versatility beyond human blood [6] [3].
Performance Across Host Taxa
The following table summarizes the detection efficacy of the nUPDx assay across different animal groups, as validated against infections confirmed by microscopy or conventional PCR.
Table 1: nUPDx Performance Across Animal Species
| Host Category | Samples Tested (Positive by other methods) | nUPDx Confirmed Infections | Key Findings | Commonly Detected Parasites |
|---|---|---|---|---|
| Mammals | 32 | 24 | Detected several undetected coinfections [6]. | Apicomplexans (e.g., Babesia spp.), nematodes [6] |
| Birds | 13 | 6 | Identified 4 previously undetected apicomplexans in cloacal samples [6] [3]. | Apicomplexans [6] |
| Reptiles | 2 | 1 | Highlights need for further validation in reptile species [6]. | Apicomplexans [6] |
| Whole Parasites | 10 (worms, arthropods) | 10 | Identified all to genus/family level; corrected one morphological misidentification [6]. | Helminths, arthropods [6] |
Advantages in Diagnostic and Surveillance Contexts
- Detection of Co-infections and Unexpected Pathogens: The broad-paneling capability of nUPDx allows for the identification of mixed infections with genetically dissimilar parasites (e.g., helminths and apicomplexans) in a single run, which is a common scenario in wildlife and domestic animals [6] [39].
- Pathogen Discovery: The assay detected Babesia sp. infections in five samples that were negative by other diagnostic approaches and identified four previously undetected apicomplexans in bird cloacal samples [6] [3].
- Methodological Validation: When applied to whole parasite specimens, nUPDx successfully identified all to the genus or family level and detected an instance where a morphological identification was incorrect, showcasing its utility in validating and refining taxonomic classifications [6].
Detailed Experimental Protocols
This section provides a standardized protocol for applying the nUPDx assay to animal-derived samples, incorporating specifics for varied sample types.
Sample Collection and Preservation
Proper sample handling is critical for downstream analysis. Recommendations vary by sample type.
Table 2: Sample Collection and Preservation Guidelines
| Sample Type | Collection Method | Preservation for Molecular Analysis | Notes |
|---|---|---|---|
| Blood | Venipuncture | -20°C or -80°C [6] | |
| Tissues | Necropsy | -20°C or -80°C [6] | |
| Feces/Scats | Non-invasive collection or from carcasses [40] | -20°C for long-term storage [40]. Room temperature if analyzed <24h [40]. | Freezing can decrease detection of some temperature-sensitive larvae [40]. |
| Whole Parasites | Collected from gut content or feces [40] | 70% ethanol; fixed in 70% ethanol and stored at -80°C [6] [39]. | Place fresh worms in warm saline to relax tissues before preservation [40]. |
Core nUPDx Wet-Lab Protocol
Procedure:
- DNA Extraction: Extract genomic DNA from samples (e.g., ~0.12g of feces, blood, or tissue lysates) using a commercial soil or tissue kit, with mechanical lysis (e.g., using a Precellys homogenizer) to ensure thorough disruption [41].
- First Restriction Digestion (D1): Digest the total DNA extract with PstI restriction enzyme. This target a cut site within the human 18S rDNA region captured by the outer primers, thereby reducing amplifiable host DNA [10].
- First (Outer) PCR:
- Primers: Use pan-eukaryotic outer primers flanking the ~200bp 18S rDNA target.
- Cycle Conditions: Initial denaturation at 95°C for 5 min; followed by 35 cycles of 95°C for 30s, 54°C for 30s, 68°C for 1 min; final extension at 68°C for 1 min [39].
- Second Restriction Digestion (D2): Digest the first PCR product with BsoBI (or alternatively, BamHI-HF). This enzyme cuts at sites present in vertebrate 18S rDNA but absent in many blood parasites, further depleting host amplicons [10].
- Second (Nested) PCR:
- Library Preparation & Sequencing: Purify the final amplicons, normalize concentrations, and pool for sequencing on an Illumina platform (e.g., MiSeq) using a 500-cycle kit [39].
Bioinformatic Analysis Workflow
A typical analysis pipeline involves:
- Demultiplexing: Assign sequences to samples based on unique barcodes.
- Quality Filtering & Trimming: Use tools like Trimmomatic or Cutadapt to remove low-quality bases and adapter sequences.
- ASV Inference: Denoise sequences to generate Amplicon Sequence Variants (ASVs) using DADA2 or UNOISE3, which corrects sequencing errors to identify biologically real sequences [41].
- Taxonomic Assignment: Classify ASVs against a curated parasitological database (e.g., SILVA, PR2) using a classifier like naïve Bayes as implemented in QIIME2 or DADA2.
- Quantification and Reporting: Report the relative abundance of parasitic taxa based on read counts per ASV.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for nUPDx Implementation
| Reagent / Material | Function / Application | Specific Example / Note |
|---|---|---|
| Pan-Eukaryotic 18S Primers | Amplification of target rDNA region from diverse parasites. | Nested primer sets designed to flank a region with vertebrate-specific restriction sites [10]. |
| PstI & BsoBI Restriction Enzymes | Selective digestion of host (vertebrate) 18S rDNA to reduce background. | Critical for enhancing sensitivity by depleting host DNA prior to PCR amplification [10]. |
| High-Fidelity DNA Polymerase | Accurate amplification of template DNA for sequencing. | Reduces PCR-derived sequencing errors. |
| NucleoSpin Soil Kit | DNA extraction from complex samples like feces and tissues. | Includes inhibitors removal steps; mechanical lysis recommended [41]. |
| Illumina MiSeq System | Deep amplicon sequencing of pooled libraries. | Standard for high-throughput sequencing; enables multiplexing of hundreds of samples [6] [39]. |
| Curated Parasite Reference Database (e.g., SILVA) | Taxonomic classification of sequenced ASVs. | Essential for accurate species-level identification; requires comprehensive parasite sequences [41] [42]. |
Overcoming Challenges: Troubleshooting and Data Analysis for nUPDx
In universal parasite diagnostic (nUPDx) deep-amplicon sequencing research, the quality of library preparation directly determines the success of detecting and differentiating parasitic pathogens. The nUPDx assay, which relies on PCR amplification of the 18S rDNA gene followed by deep sequencing, is particularly susceptible to common preparation pitfalls that can compromise sensitivity and specificity [3] [5]. Issues such as low library yield can obscure detection of low-abundance parasites, while adapter dimer formation and amplification bias can generate false negatives or inaccurate representations of parasite community structure. These challenges are especially pronounced in diagnostic contexts where sample inputs may be limited and host DNA contamination can drastically reduce the proportion of parasite-derived reads [5]. This application note details the major pitfalls in NGS library preparation for deep-amplicon sequencing and provides optimized protocols specifically adapted for parasite diagnostics to ensure reliable and sensitive detection of parasitic infections.
Problem 1: Low Library Yield – Causes and Solutions
Low library yield presents a significant challenge in nUPDx sequencing, potentially reducing detection sensitivity for rare parasites or low-level infections. This issue is particularly critical in diagnostic applications where maximum sensitivity is required.
Table 1: Troubleshooting Low Library Yield
| Cause of Low Yield | Mechanism of Yield Loss | Corrective Action for nUPDx |
|---|---|---|
| Poor Input Quality/Contaminants | Enzyme inhibition from residual salts, phenol, or polysaccharides [43] | Re-purify input DNA; ensure 260/230 > 1.8; use clean columns/beads [43] |
| Inaccurate Quantification | Overestimating usable DNA concentration with UV absorbance [43] | Use fluorometric methods (Qubit) rather than NanoDrop [43] |
| Amplification Inefficiency | Suboptimal PCR due to primer mismatches or inhibitors [43] [5] | Validate primer specificity for target parasites; use high-fidelity polymerases; add BSA [5] |
| Overly Aggressive Cleanup | Excessive sample loss during size selection steps [43] | Optimize bead-to-sample ratios; avoid bead over-drying; implement "double-sided" cleanup [43] |
Adapted Protocol for High-Yield nUPDx Library Preparation
Procedure: This protocol modifies the established nUPDx approach to maximize yield for parasite detection in complex host backgrounds [5].
Input DNA Preparation:
- Extract DNA using bead-beating or enzymatic lysis optimized for diverse parasite types.
- Quantify using fluorometric methods (e.g., Qubit dsDNA HS Assay) rather than UV absorbance to avoid overestimation from contaminants [43].
- For whole blood samples, implement a host DNA depletion step or use restriction enzyme digestion to selectively digest vertebrate DNA while preserving parasite DNA [5].
First-Stage PCR (Target Amplification):
- Use nested PCR targeting the 18S rDNA gene with primers containing overhang adapters [5] [44].
- Modified Approach: Incorporate Illumina barcodes and adapters directly during PCR amplification to eliminate a separate library preparation step, reducing turnaround time and sample loss [5].
- Reaction Setup:
- High-fidelity DNA polymerase (e.g., KAPA HiFi HotStart ReadyMix)
- 10-50 ng genomic DNA (or 5 µL if concentration is low)
- Primer set with overhang adapters (0.5 µM each)
- Cycling Conditions: 95°C for 3 min; 35 cycles of 98°C for 20s, 60°C for 30s, 72°C for 30s; final extension at 72°C for 5 min.
Purification and Cleanup:
- Purify PCR products using magnetic beads (e.g., AMPure XP) at a 0.8x ratio to remove primers and primer dimers.
- Elute in nuclease-free water or low TE buffer. Avoid over-drying beads, which can lead to poor resuspension and significant DNA loss [43].
Figure 1: Optimized nUPDx Library Prep Workflow for Maximum Yield
Problem 2: Adapter Dimers and Contamination – Causes and Solutions
Adapter dimers form when sequencing adapters ligate to themselves rather than to target DNA fragments. These artifacts can dominate sequencing runs, drastically reducing reads from target parasite DNA and compromising detection sensitivity.
Table 2: Troubleshooting Adapter Dimers and Contamination
| Cause of Adapter Dimers | Impact on nUPDx | Prevention Strategy |
|---|---|---|
| Excess Adapters | Adapter-dominated sequencing; reduced parasite reads [43] | Titrate adapter:insert molar ratios; implement cleanup after ligation [43] |
| Inefficient Ligation | Poor adapter incorporation; increased free adapters [43] | Ensure fresh ligase/buffer; optimal temperature (~20°C) [43] |
| Inadequate Size Selection | Failure to remove dimer artifacts before sequencing [43] | Implement double-sided size selection (e.g., 0.55x/0.8x bead ratios) [43] |
| Low Input DNA | Increased ratio of adapter-to-target molecules [43] | Use carrier RNA for very low input samples; minimize PCR cycles [43] |
Optimized Protocol for Adapter Dimer Prevention in nUPDx
Procedure: This protocol employs a modified nested PCR approach that incorporates full adapters during amplification, eliminating the separate ligation step that often generates dimers [5].
Adapter-Integrated Amplification:
- Design first-stage PCR primers with overhangs that contain complementary sequences to Illumina adapters.
- Use a second-stage PCR to attach full dual indexes and sequencing adapters.
- This "PCR-only" approach bypasses traditional ligation, dramatically reducing adapter dimer formation [5].
Size Selection Optimization:
- Implement a double-sided SPRIselect bead cleanup between PCR stages:
- Lower cutoff: Use 0.55x ratio to remove small fragments and primer dimers.
- Upper cutoff: Use 0.8x ratio to retain target amplicons while excluding large non-specific products.
- Resuspend beads thoroughly by pipetting or vortexing to prevent sample loss.
- Implement a double-sided SPRIselect bead cleanup between PCR stages:
Rigorous QC Assessment:
- Analyze final libraries using BioAnalyzer, TapeStation, or fragment analyzer.
- Look for a clean peak in the expected size range (e.g., 300-500 bp for nUPDx) and absence of the ~70-90 bp adapter dimer peak [43].
- Quantify using qPCR-based methods (e.g., Kapa Library Quantification) for accurate measurement of amplifiable library molecules.
Problem 3: Amplification Bias and Uneven Coverage – Causes and Solutions
Amplification bias occurs when certain template molecules are preferentially amplified over others, leading to uneven coverage that can distort parasite representation and miss low-abundance species. This is particularly problematic in nUPDx when detecting mixed parasite infections or performing relative quantitation.
Table 3: Troubleshooting Amplification Bias
| Source of Bias | Effect on nUPDx Results | Mitigation Approach |
|---|---|---|
| Over-Amplification | Skews representation toward dominant sequences; increases duplicate reads [43] | Use minimal PCR cycles; optimize cycle number for each sample type [43] |
| Primer Mismatches | Differential amplification of parasite species with 18S sequence variations [5] | Use degenerate primers; validate against diverse parasite sequences [5] |
| GC Content Bias | Under-representation of GC-rich parasite genomes [45] | Use polymerases optimized for GC-rich templates; add PCR enhancers [45] |
| Enzyme Inhibitors | Carryover contaminants cause partial inhibition and skewed amplification [43] | Ensure thorough cleanup post-extraction; include BSA or other enhancers [43] |
Bias Minimization Protocol for nUPDx Applications
Procedure: This protocol outlines specific modifications to reduce amplification bias in parasite detection assays.
PCR Optimization:
- Cycle Determination: Use the minimum number of PCR cycles needed for adequate library yield. Typically 25-35 cycles for nUPDx, but should be empirically determined [43].
- Polymerase Selection: Employ high-fidelity polymerases with minimal sequence bias (e.g., KAPA HiFi, Q5 Hot Start).
- Reaction Enhancers: Include 1M betaine or 1x Q-Solution to mitigate GC bias, particularly important for diverse parasite genomes.
Primer Design Strategy:
- Use broadly conserved 18S rDNA primers with strategic degeneracy to cover diverse parasite taxa.
- Validate in silico against available sequences of target parasites to identify potential mismatches.
- For the modified nUPDx approach, incorporate Illumina adapter sequences directly into the primer design to eliminate ligation bias [5].
Library Normalization and Pooling:
- Precisely quantify final libraries using qPCR-based methods.
- Normalize to equal molarity before pooling to ensure balanced representation of samples.
- For nUPDx, include non-template controls and positive controls (with known parasite DNA) in each run to monitor amplification efficiency and potential bias.
The Scientist's Toolkit: Essential Reagents and Solutions
Table 4: Key Research Reagent Solutions for nUPDx Library Preparation
| Reagent/Category | Specific Examples | Function in nUPDx Workflow |
|---|---|---|
| High-Fidelity DNA Polymerase | KAPA HiFi HotStart, Q5 Hot Start | Accurate amplification of 18S rDNA target with minimal bias [5] |
| Magnetic Beads | AMPure XP, SPRIselect | Size selection and cleanup; removal of primers, adapter dimers, and contaminants [43] |
| Quantification Kits | Qubit dsDNA HS Assay, Kapa Library Quantification Kit | Accurate measurement of input DNA and final library concentration [43] |
| Nested PCR Primers | Custom 18S rDNA primers with Illumina adapter overhangs | Specific amplification of parasite DNA; incorporation of sequencing adapters [5] |
| Library Preparation Kit | AmpliSeq for Illumina, Illumina DNA Prep | Streamlined library preparation in optimized, validated formulations [45] [46] |
The success of universal parasite diagnostic sequencing hinges on overcoming critical pitfalls in NGS library preparation. By implementing the detailed protocols and troubleshooting guides provided herein—particularly the modified nUPDx approach that incorporates adapters during PCR amplification—researchers can significantly improve library yield, eliminate adapter dimers, and minimize amplification bias. These optimizations collectively enhance the sensitivity and accuracy of parasite detection, which is essential for both clinical diagnostics and wildlife surveillance where unexpected or low-abundance pathogens must be reliably identified. The strategies outlined support the development of more robust and cost-effective parasite diagnostic sequencing, with the modified nUPDx approach reducing costs from approximately $40 to $11 per sample while maintaining high sensitivity [5].
Targeted amplicon deep sequencing (TADS) has emerged as a powerful methodology for sensitive detection and characterization of parasitic pathogens in complex biological samples. This protocol details the application of two distinct bioinformatics pipelines, AmpSeqR and DADA2, for processing raw FASTQ files to high-fidelity haplotype calls, specifically within the context of universal parasite diagnostic (nUPDx) research. The nUPDx approach, which relies on deep-amplicon sequencing of the 18S rDNA gene, enables the detection and differentiation of diverse blood-borne parasites such as Plasmodium spp., Babesia spp., kinetoplastids, and filarial nematodes from clinical specimens, often in a single assay [6] [5] [10]. We provide step-by-step methodologies, benchmark performance metrics, and a curated toolkit of research reagents to equip researchers and drug development professionals with the resources to implement these robust, reproducible pipelines in their own diagnostic and surveillance workflows.
The molecular diagnosis of parasitic infections has traditionally been challenged by the need for multiple species-specific assays, which can miss unknown or co-infecting pathogens. The nested Universal Parasite Diagnostic (nUPDx) approach addresses this by using a pan-eukaryotic PCR targeting the 18S rDNA locus, followed by deep-amplicon sequencing. This method has demonstrated a limit of detection (LOD) as low as 0.58 Plasmodium falciparum parasites/µL of blood, making it comparable to real-time PCR assays in sensitivity [5] [10]. A key innovation in nUPDx is the incorporation of restriction enzyme digestion steps that selectively cleave vertebrate (host) 18S rDNA sequences, which are absent in the target parasites. This enrichment strategy drastically increases the proportion of parasite-derived reads, enabling the detection of minor clones in mixed samples [10].
The transformation of raw sequencing reads into reliable biological insights requires sophisticated bioinformatics pipelines to distinguish true haplotypes from artifacts introduced during PCR amplification and sequencing. This application note focuses on two such pipelines:
- AmpSeqR: An end-to-end R package specifically tailored for amplicon deep sequencing data, with a focus on infectious diseases [47].
- DADA2: A widely-used R package that models and corrects sequencing errors to infer amplicon sequence variants (ASVs) [48].
The following sections provide a detailed guide to implementing these pipelines, from experimental design and data preprocessing to downstream analysis and visualization, all framed within the practical requirements of nUPDx research.
Pipeline Comparison and Performance Benchmarks
The choice between AmpSeqR and DADA2 depends on the project's specific needs, such as the desired level of automation, the need for specialized post-processing, or integration with existing workflows. The table below summarizes the key characteristics of each pipeline to aid in this decision.
Table 1: Comparative overview of the AmpSeqR and DADA2 pipelines.
| Feature | AmpSeqR | DADA2 |
|---|---|---|
| Primary Focus | End-to-end analysis with emphasis on infectious disease data [47] | General-purpose amplicon analysis, widely used in microbiome studies [48] |
| Typical Input | Raw paired-end FASTQ, sample barcodes, amplicon details [47] | Demultiplexed, primer-trimmed paired-end FASTQ files [48] |
| Core Analysis Engine | Integrates DADA2 for ASV inference and adds custom functions [47] | Proprietary error modeling and ASV inference algorithm [48] |
| Key Distinguishing Features | Automated report generation; integrated post-processing for noise filtering (chimeras, indels); data visualization [47] | High-resolution ASV inference; flexible, modular workflow; large user community [48] |
| Report Generation | Automatically generates a comprehensive Rmarkdown report [47] | Requires manual scripting for report generation |
| Best Suited For | Projects requiring a streamlined, all-in-one solution for pathogen amplicon data | Projects requiring a highly customizable, modular workflow, or for microbiome data |
Performance benchmarks are critical for planning and resource allocation. AmpSeqR has been benchmarked on real-world datasets, with runtimes varying based on data size and computational resources.
Table 2: Runtime benchmarks for major AmpSeqR functions across different datasets [47].
| AmpSeqR Function | Small Dataset | Medium Dataset | Large Dataset |
|---|---|---|---|
| Data Pre-processing | 15 minutes | 45 minutes | 2 hours |
| ASV Estimation | 30 minutes | 1.5 hours | 4 hours |
| Data Post-processing | 10 minutes | 25 minutes | 1 hour |
| Report Generation | 5 minutes | 15 minutes | 30 minutes |
Experimental Protocols
Universal Parasite Diagnostic (nUPDx) Wet-Lab Protocol
The bioinformatics pipelines described later process data generated from a specific nested PCR-based assay.
- Principle: The assay uses two sets of pan-eukaryotic primers targeting the 18S rDNA. Restriction enzyme sites present in vertebrate (host) DNA but absent in target parasites are exploited to digest host DNA between PCR rounds, enriching the sample for parasite-derived amplicons [10].
- Key Steps:
- DNA Extraction: Extract total DNA from blood, tissue, or other biological specimens.
- First Restriction Digestion (D1): Digest the total DNA extract with PstI to reduce amplifiable host DNA.
- First (Outer) PCR: Amplify the target 18S rDNA region using the outer primer set.
- Second Restriction Digestion (D2): Digest the first PCR product with BsoBI (or BamHI) to further reduce any residual host amplicons.
- Second (Nested) PCR: Amplify the inner ~200 bp target from the digested first PCR product. This step can incorporate Illumina barcodes and adapters, making the amplicons ready for sequencing without a separate library preparation step, reducing cost and time [5].
- Sequencing: Pool and sequence the final amplicons on an Illumina MiSeq or similar platform.
Bioinformatics Protocol I: The AmpSeqR Pipeline
AmpSeqR is designed as a comprehensive workflow that starts from raw sequencing reads and finishes with a final report.
Step 1: Data Pre-processing
- Demultiplexing: Assign raw paired-end FASTQ reads to individual samples based on their unique oligonucleotide barcodes.
- Trimming: Trim sample barcodes and target amplicon primer sequences. This step uses the
ShortReadandBiostringsR packages [47].
Step 2: Amplicon Sequence Variants (ASVs) Estimation
- Quality Filtering and Trimming: Use
DADA2::filterAndTrimto remove low-quality reads. Parameters such astruncLen(e.g., 240 for forward, 160 for reverse reads) are set based on quality profiles [48]. - Error Model Learning: Execute
DADA2::learnErrorsto create a sample-specific error model. - Dereplication and ASV Inference: Run
DADA2::derepFastqandDADA2::dadato identify the core haplotypes. - Read Merging: Use
DADA2::mergePairsto combine paired-end reads. AmpSeqR provides an option to process non-overlapping pairs [47]. - Automatic Downsampling: To reduce computation time, AmpSeqR can automatically downsample each sample/amplicon combination to a sufficient depth (e.g., 10,000 reads) for detecting minor clones as low as 0.1% [47].
Step 3: Data Post-processing
- Chimera Removal: Identify and remove chimeric reads formed during PCR.
- Variant Filtration: Filter haplotypes based on parameters including:
- Haplotype frequency (ultra-rare variants are discarded).
- Sequence similarity against a reference sequence for each amplicon marker.
- Variant heterozygosity and minor allele frequency (MAF) across the dataset [47].
Step 4: Data Visualization and Reporting
- AmpSeqR automatically generates plots and a comprehensive Rmarkdown report, facilitating the inclusion of results into publications [47].
Bioinformatics Protocol II: The DADA2 Pipeline
The DADA2 pipeline is a modular, community-standard workflow that offers fine-grained control at each step.
Step 1: Primer Removal with cutadapt
- Before starting in DADA2, remove primers using a tool like
cutadapt. This is critical for accurate downstream processing [49]. - Example Command in R:
Step 2: Quality Filtering and Trimming
- Inspect read quality profiles using
plotQualityProfile(fnFs[1:2])andplotQualityProfile(fnRs[1:2])to inform trimming parameters [48]. - Filter and trim reads using
filterAndTrim. Typical parameters includetruncLen=c(240, 160),maxEE=c(2,2),truncQ=2, andrm.phix=TRUE[48].
Step 3: Core DADA2 Algorithm
- Learn Error Rates: Execute
errF <- learnErrors(filtFs, multithread=TRUE)and similarly for reverse reads. Visualize the error models withplotErrors(errF, nominalQ=TRUE)[48]. - Dereplication: Run
derepFastqto combine identical reads. - Sample Inference: Apply the core
dadaalgorithm to infer true ASVs in each sample. - Merge Paired-end Reads: Use
mergePairsto create the full-length amplicon sequences.
Step 4: Construct ASV Table and Remove Chimeras
- Create a sequence table with
makeSequenceTable. - Remove chimeras with
removeBimeraDenovo, which is essential for avoiding false positive haplotypes [48].
Step 5: Taxonomic Assignment (Optional for nUPDx)
- While nUPDx often relies on BLAST against custom parasite databases, DADA2 provides
assignTaxonomy(andaddSpecies) for classification against standard 16S/18S databases [48] [50].
Workflow Visualization
The following diagram illustrates the key decision points and steps in a standard amplicon analysis pipeline, integrating both AmpSeqR and DADA2 concepts.
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of the nUPDx assay and its associated bioinformatics pipelines relies on a set of key reagents, software, and reference databases.
Table 3: Essential research reagents, software, and data resources for nUPDx and amplicon analysis.
| Category | Item | Function / Application |
|---|---|---|
| Wet-Lab Reagents | Pan-eukaryotic 18S rDNA Primers | For nested PCR amplification of the target locus from a wide range of parasites [10]. |
| Restriction Enzymes (PstI, BsoBI) | For selective digestion of host (vertebrate) 18S rDNA between PCR rounds to enrich parasite DNA [10]. | |
| Bioinformatics Software | R and RStudio | The foundational computing environment for running the AmpSeqR and DADA2 pipelines [48] [49]. |
| AmpSeqR R Package | An integrated pipeline for amplicon data from FASTQ to final report, ideal for pathogen-focused studies [47]. | |
| DADA2 R Package | A modular, high-resolution pipeline for inferring amplicon sequence variants (ASVs) [48]. | |
| cutadapt | A software tool for finding and removing primer sequences from amplicon reads; used prior to DADA2 [49]. | |
| Reference Data | Custom Parasite 18S rDNA Database | A curated BLAST database for assigning taxonomy to ASVs from clinical specimens; crucial for nUPDx [6] [20]. |
| IDTAXA-formatted Databases | Formatted taxonomy files (e.g., for nematode ITS2) for use with alternative classification methods [49]. | |
| Positive Control | ZymoBIOMICS Microbial Community DNA Standard | A defined mock community of genomic DNA used to validate assay performance and bioinformatics accuracy [51]. |
The integration of robust wet-lab assays like nUPDx with sophisticated bioinformatics pipelines such as AmpSeqR and DADA2 provides a powerful framework for universal parasite detection. The AmpSeqR pipeline offers a streamlined, all-in-one solution specifically optimized for infectious disease data, automatically handling complex post-processing and reporting. In contrast, the DADA2 pipeline provides a flexible, modular foundation that allows for extensive customization. Both pipelines enable researchers to move confidently from raw sequencing data to high-resolution haplotype calls, detecting minor clones at frequencies as low as 0.1%—a critical capability for diagnosing mixed infections and tracking pathogen dynamics. By adhering to the detailed protocols and utilizing the toolkit provided herein, research and drug development teams can reliably implement these methods to advance studies in parasite ecology, evolution, and diagnosis.
Distinguishing True Minor Clones from Sequencing Errors and Chimeric Reads
Within the framework of universal parasite diagnostic (nUPDx) deep-amplicon sequencing research, a primary analytical challenge is the accurate discrimination between true low-frequency minor clones and artifacts generated by the sequencing process itself. The nUPDx assay, which employs a nested PCR targeting the 18S rDNA gene coupled with restriction enzyme digestion of host DNA, provides a powerful tool for detecting blood-borne parasites such as Plasmodium, Babesia, kinetoplastids, and filarial nematodes with a sensitivity comparable to real-time PCR (approximately 0.58 parasites/μL for Plasmodium falciparum) [16]. However, the full potential of this assay in characterizing complex, multi-clone infections and detecting rare variants is contingent upon robust bioinformatic techniques to mitigate sequencing errors and chimeric reads. This application note details standardized protocols and analytical frameworks to address this critical issue, ensuring the reliability of minority clone detection in clinical and research settings.
Critical Data and Performance Metrics
Table 1: Performance Metrics of Amplicon Sequencing for Minor Clone Detection
| Parameter | Performance in High-Quality Samples | Performance at Low Density/Read Count | Citation |
|---|---|---|---|
| Limit of Detection (LOD) | ~0.58 P. falciparum/μL (nUPDx) | N/A | [16] |
| Minor Clone Detection Threshold | Robust detection >1% frequency | Reduced sensitivity & precision <1% frequency | [52] |
| Accuracy of Major vs. Minor Haplotype Call | 90% (at ≥30 genomes/μL) | 61% (at <5 genomes/μL) | [53] [54] |
| Recommended Sequencing Coverage | >10,000 reads/amplicon | <100 reads/amplicon leads to higher false positives | [52] |
| Sequencing Error Rate (Post-Trim) | 0.38% - 0.50% per nucleotide | Increases significantly towards read ends | [52] [54] |
Table 2: Comparison of Bioinformatic Tools for Haplotype Calling
| Tool Name | Primary Approach | Reported Strengths / Notes | Citation |
|---|---|---|---|
| PASEC | Distance- and abundance-based error correction | Tailored for specific amplicons (e.g., CSP, SERA2); can identify indels | [54] |
| DADA2 | Divisive amplicon denoising algorithm | Models and corrects Illumina amplicon errors | [54] |
| HaplotypR | SNP-based clustering and haplotype filtering | Includes quality checks based on sample replicates | [52] [54] |
| SeekDeep | Processing pipeline for targeted amplicons | Designed to handle samples from diverse populations | [54] |
Experimental Protocols
Protocol for nUPDx Assay with Enhanced Sensitivity
This protocol is designed for the sensitive detection of blood parasites while mitigating host DNA interference [10] [16].
Step 1: DNA Extraction and Primary Restriction Digestion (D1)
- Extract total DNA from patient blood samples using a commercial nucleic acid extraction kit.
- Perform the first restriction enzyme digestion (Digest 1) on the total DNA extract using PstI. This enzyme targets a cut site within the human 18S rDNA sequence, thereby selectively digesting host DNA and enriching the relative proportion of amplifiable parasite DNA.
Step 2: Primary Nested PCR
- Perform the first PCR amplification using outer pan-eukaryotic primers that flank the target ~200-bp region of the 18S rDNA gene.
- Primer Sequences: As described by Flaherty et al. [10].
- Cycling Conditions: Initial denaturation at 95°C for 5 min; followed by 25 cycles of 95°C for 30s, 55°C for 30s, 72°C for 45s; and a final extension at 72°C for 7 min.
Step 3: Secondary Restriction Digestion (D2)
- Perform the second restriction enzyme digestion (Digest 2) on the product of the first PCR using BsoBI (or alternatively, BamHI-HF). This step further reduces any residual amplifiable host-derived 18S rDNA amplicons, which contain restriction sites absent in the target parasites [10].
Step 4: Secondary Nested PCR with Adapters
- Perform the second PCR using inner pan-eukaryotic primers that bind within the initial amplicon.
- Primer Modification: Incorporate Illumina adapter overhangs into these inner primers to facilitate direct library preparation [16].
- Cycling Conditions: Use 25-30 cycles with similar temperatures as the first PCR.
Step 5: Sequencing
- Purify the final amplicons and quantify the library.
- Sequence on an Illumina MiSeq platform using a 500-cycle or 600-cycle reagent kit to generate paired-end reads.
Protocol for Validating Minor Clones Using Mock Communities
This methodology is critical for establishing the limit of detection and error rates of your assay [52] [54].
Step 1: Sample Preparation
- Create mock mixtures of DNA from known culture-adapted parasite lines (e.g., P. falciparum strains 3D7 and HB3).
- Mix the DNA in defined proportions to simulate polyclonal infections with known minority clone frequencies (e.g., 1%, 5%, 10%).
- Spike the parasite DNA mixtures into human genomic DNA (e.g., 10 ng) to mimic the clinical sample matrix.
Step 2: Library Preparation and Sequencing
- Process these mock samples alongside clinical unknowns using the nUPDx protocol described above.
- Include technical replicates (duplicate or triplicate PCRs/sequencing for each sample) to distinguish consistent true variants from stochastic errors.
Step 3: Data Analysis and Threshold Determination
- Process the sequencing data through your chosen bioinformatic pipeline (e.g., PASEC, DADA2).
- Compare the detected haplotypes and their frequencies to the known input composition of the mock samples.
- Establish a minimum read count threshold (e.g., >100 reads per amplicon) and a minimum frequency threshold (e.g., >1%) for reporting minor clones, based on the empirical validation data [52].
Workflow Visualization
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for nUPDx
| Research Reagent / Tool | Function / Application | Specific Example / Note |
|---|---|---|
| Pan-Eukaryotic 18S rDNA Primers | Amplification of target region from diverse parasites while retaining vertebrate-specific restriction sites. | Nested primer sets (outer and inner) as per Flaherty et al. [10]. |
| Restriction Enzymes (PstI, BsoBI) | Selective digestion of host-derived 18S rDNA amplicons to enrich parasite DNA signal. | PstI for first digest (D1) on gDNA; BsoBI for second digest (D2) on primary PCR product [10]. |
| Illumina-Compatible Adapter Primers | Integration of sequencing adapters during PCR, eliminating separate library prep. | Inner nested primers modified with Illumina overhangs [16]. |
| Mock Parasite DNA Communities | Assay validation, determining LOD, and establishing error rates for minor clone detection. | Defined mixtures of culture-adapted Plasmodium strains [52] [54]. |
| Bioinformatic Pipelines (e.g., DADA2, HaplotypR) | Denoising raw sequence data, correcting errors, and calling haplotypes. | Use of multiple algorithms can improve variant calling accuracy [54]. |
Optimizing Coverage and Read Depth for Sensitive Minority Variant Detection
Within the framework of universal parasite diagnostic (nUPDx) deep-amplicon sequencing research, the reliable detection of minority variants is paramount. These low-frequency genetic variants, which can indicate emerging drug resistance or mixed parasite infections, are often obscured by technical noise inherent to sequencing workflows [55]. This application note provides detailed protocols and data-driven guidelines for optimizing coverage and read depth to enhance the sensitivity and specificity of minority variant detection in parasitic pathogens using the nUPDx platform.
The nUPDx assay, which employs PCR amplification of the 18S rDNA locus followed by deep-amplicon sequencing, provides a universal detection method for blood-borne parasites [3] [21]. Its adaptation for minority variant detection requires careful optimization of wet-lab procedures and sequencing parameters to distinguish true biological variants from artifacts introduced during library preparation and sequencing [55] [5].
Key Parameters for Minority Variant Detection
Table 1: Key Experimental Parameters Affecting Minority Variant Detection
| Parameter | Impact on Sensitivity | Optimal Range/Value | Supporting Evidence |
|---|---|---|---|
| Sequencing Depth | Directly influences ability to detect low-frequency variants; higher depth increases confidence. | >1000x per amplicon | Simulation data indicates high depth crucial for variants <1% [55] |
| Limit of Detection (LOD) for Minority Variants | Defines the lowest variant allele frequency (VAF) that can be reliably called. | 10% (for validated assays like Deeplex Myc-Lep) | Empirically determined for M. leprae [56] |
| Viral Copy Number Input | Lower template copies increase stochastic PCR effects and reduce variant detection. | 80-3000 genome copies (LOD for sequencing success) | Correlation established between input and success [56] |
| Polymerase Fidelity | High-fidelity enzymes reduce introduction of errors during amplification that mimic true variants. | Use of Q5 Hot Start High-Fidelity DNA Polymerase | Standard in optimized protocols to minimize noise [57] |
| Amplicon Size | Affects amplification efficiency and uniformity of coverage. | Varies; long amplicons (~3kb) successful for RSV | Balanced efficiency and coverage [58] |
Table 2: Impact of Sequencing Depth on Ground Truth Mutation Detection
| Sequencing Depth | Detection Rate of Ground Truth Mutations | Key Observations | Source |
|---|---|---|---|
| Low Depth | Low | High false-negative rate for low-frequency variants; insufficient coverage for confident calling. | Simulation Study [55] |
| High Depth | High | Enables confident identification of low-frequency variants down to the defined LOD of the assay. | Simulation Study [55] |
| Saturated Depth | Diminishing Returns | Beyond a certain point, increased cost yields minimal gains in detection sensitivity. | Simulation Study [55] |
Experimental Protocols
Protocol 1: Optimized nUPDx Library Preparation for Minority Variants
This protocol is modified from the original nUPDx assay to enhance sensitivity for minority variants while reducing cost and turnaround time [5].
Materials:
- Primers: nUPDx 18S rDNA primers (modified to include Illumina barcodes and adapters) [5]
- Enzymes: High-fidelity DNA polymerase (e.g., Q5 Hot Start High-Fidelity)
- Restriction Enzymes: For selective host DNA digestion (e.g., HhaI)
- Clean-up Reagents: AMPure XP beads
- Sample Input: 200 μL of whole blood preserved in EDTA
Procedure:
- DNA Extraction: Extract genomic DNA from 200 μL of EDTA-blood using a commercial kit (e.g., QIAamp DNA Mini Kit) on a QIAcube instrument, with modifications for no tip reuse to prevent cross-contamination [24].
- Nested PCR with Integrated Barcoding:
- First PCR: Perform the initial amplification using eukaryotic-specific 18S rDNA primers.
- Host DNA Depletion: Digest the primary PCR product with restriction enzymes (e.g., HhaI) that selectively cleave host-derived 18S rDNA sequences, thereby enriching parasite DNA [24] [21].
- Second PCR (Barcoding): Use barcoded primers for the nested amplification. These primers incorporate full Illumina adapter sequences, making the final amplicons immediately ready for sequencing and eliminating a separate library preparation step [5].
- Library Clean-up and Normalization: Purify the final PCR products using AMPure XP beads at a 0.5x ratio to remove primers and short fragments. Quantify amplicons using a fluorescence-based method (e.g., QuantiFluor dsDNA System) and pool equimolar amounts of each library [58].
- Sequencing: Sequence the pooled library on an Illumina MiSeq or similar platform. Aim for a minimum depth of 1000x coverage per amplicon to ensure sensitive detection of minority variants.
Protocol 2: In-silico Optimization Using GENOMICON-Seq
This simulation-based protocol allows for the benchmarking of variant-calling parameters and the assessment of new laboratory protocols in silico before wet-lab experimentation, saving time and resources [55].
Materials:
- Software: GENOMICON-Seq (Docker-based package available at https://github.com/Rounge-lab/GENOMICON-Seq)
- Reference Genome: FASTA file of the target parasite genome(s).
- Mutation Profile: Defined list of ground truth mutations or a mutational signature (e.g., COSMIC SBS signatures).
Procedure:
- Define Input Sample: Provide a FASTA file of the genome(s) of interest and set the copy number for each to model different pathogen loads.
- Insert Ground Truth Mutations: Introduce low-frequency mutations using one of GENOMICON-Seq's modes:
- Deterministic Mode: For precise control, specify the exact number and distribution of mutations across genome copies to model desired VAFs.
- SBS-Mimicry Mode: To model realistic mutation patterns, use pre-defined mutational signatures (e.g., from COSMIC) to assign mutations [55].
- Simulate Technical Noise: Configure the simulation to model key sources of noise:
- Set the polymerase error rate for the PCR simulation.
- For probe-capture simulations (WES), define probe sequences and overlap rules.
- Specify sequencing depth and introduce sequencing platform-specific error profiles.
- Generate and Analyze Output: Run the simulation to generate FASTQ files and a summary file tracking all ground truth and error-derived mutations. Use these outputs to test and refine your variant-calling pipeline's accuracy and establish optimal filtering thresholds (e.g., minimum allele frequency, read depth) for detecting true low-frequency variants [55].
Workflow and Relationship Diagrams
Figure 1: Optimized nUPDx wet-lab workflow for minority variant detection, incorporating integrated barcoding and host DNA depletion.
Figure 2: Key parameters and their interrelationships influencing detection sensitivity.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions
| Item | Function/Application | Specific Example |
|---|---|---|
| High-Fidelity DNA Polymerase | Reduces PCR-induced errors that confound minority variant detection. | Q5 Hot Start High-Fidelity DNA Polymerase (NEB) [57] |
| Restriction Enzymes | Selectively digests host (e.g., human) 18S rDNA amplicons, enriching parasite-derived sequences and improving signal-to-noise ratio. | HhaI [24] [21] |
| Magnetic Beads | Purifies and size-selects amplicons post-amplification; removes primer dimers and short fragments. | AMPure XP Beads (Beckman Coulter) [57] [58] |
| Barcoded Primers | Enables multiplexing of samples by adding unique indices during PCR; integrated adapters streamline library prep. | Illumina-compatible barcoded primers [57] [5] |
| In-silico Simulation Tool | Benchmarks variant calling and optimizes wet-lab protocols computationally before testing. | GENOMICON-Seq [55] |
Addressing Primer-Template Mismatches to Broaden Parasite Species Coverage
Universal parasite diagnostic (nUPDx) deep-amplicon sequencing represents a transformative approach for detecting diverse blood-borne parasites from a single test. A significant challenge in designing such broad assays is addressing primer-template mismatches, which occur due to genetic diversity across parasite species and can drastically reduce detection sensitivity. This application note details the mechanisms by which mismatches impair assay performance and provides a validated, nested PCR-based protocol to broaden species coverage while maintaining high sensitivity for clinically relevant parasites, including Plasmodium spp., Babesia spp., kinetoplastids, and filarial nematodes.
The nested Universal Parasite Diagnostic (nUPDx) assay is a targeted amplicon deep sequencing (TADS) strategy designed to detect a wide spectrum of blood-borne parasites by targeting a segment of the 18S rDNA gene [16]. This assay innovatively uses restriction enzyme sites present in vertebrate (host) 18S rDNA but absent in parasites to selectively digest host DNA, thereby enriching the relative proportion of parasite-derived amplicons for sequencing [10]. This method has demonstrated a limit of detection (LOD) comparable to pathogen-specific real-time PCR assays, proving effective for malaria parasites, Babesia species, kinetoplastids, and filarial nematodes [16].
A critical hurdle in universal assay design is primer-template mismatches. These occur when the designed primer sequences are not perfectly complementary to the target genomic DNA of all intended parasite species due to natural genetic variation. Such mismatches can reduce PCR amplification efficiency by lowering the melting temperature (Tm) of the primer-template hybrid, potentially leading to false-negative results and a narrowing of effective species coverage [59]. Even a 1% base mismatch can reduce the Tm by 1.0–1.4°C, and single mismatches, depending on their position and type, can increase the cycle threshold (Ct) by more than 7.0 cycles, severely impacting detection sensitivity [59]. Addressing this is paramount for developing a truly robust universal diagnostic.
Quantitative Impact of Mismatches on PCR Efficiency
The following table summarizes key experimental findings on how primer-template mismatches affect PCR assay performance, which directly informs the design of robust nUPDx primers.
Table 1: Experimental Data on the Impact of Primer-Template Mismatches on PCR Efficiency
| Parameter | Impact on PCR | Experimental Context & Key Findings |
|---|---|---|
| General Tm Reduction | 1.0–1.4°C reduction per 1% base mismatch [59] | Fundamental thermodynamic effect of sequence divergence. |
| Single Mismatch Effect (Varied) | Ct shift range: <1.5 to >7.0 [59] | Minor Impact (<1.5 Ct): A-C, C-A, T-G, G-T mismatches.Severe Impact (>7.0 Ct): A-A, G-A, A-G, C-C mismatches. |
| Mismatch Position | Critical influence [59] | Single mismatches located >5 bp from the 3' end have a moderate effect and can often be tolerated. Mismatches at the 3' end are far more disruptive to amplification. |
| Number of Mismatches | Increased impact with count [59] | A complete blocking of the PCR reaction was observed with 4 mismatches in experimental setups. |
| nUPDx Assay Performance | LOD of 0.58 P. falciparum/μL [16] | Demonstrates that careful primer design and a nested PCR approach with host DNA digestion can achieve high sensitivity despite taxonomic diversity. |
Protocol for a Mismatch-Tolerant nUPDx Assay
This protocol outlines the Ad_UPDx method, an optimized version of nUPDx that incorporates Illumina adapters during PCR, reducing cost and turnaround time while maintaining sensitivity and broad coverage [16].
Research Reagent Solutions
Table 2: Essential Reagents and Materials for the nUPDx Assay
| Item | Function / Application | Specification / Note |
|---|---|---|
| Pan-Eukaryotic Primers | Amplification of 18S rDNA target region from both host and parasite DNA [10]. | Outer and inner primer sets are used in a nested configuration. Inner primers can be modified with Illumina adapter overhangs [16]. |
| Restriction Enzymes (e.g., BsoBI) | Selective digestion of host-derived 18S rDNA amplicons [10]. | Targets enzyme cut sites present in vertebrate 18S rDNA but absent in target parasites, enriching the parasite DNA fraction. |
| High-Fidelity DNA Polymerase | PCR amplification of the target locus. | Reduces error rates during amplification, crucial for accurate sequencing. |
| Illumina MiSeq Reagents | Next-generation sequencing of the final amplicon library. | The modified Ad_UPDx protocol makes amplicons immediately ready for MiSeq sequencing [16]. |
Step-by-Step Workflow
Step 1: Initial Restriction Digestion and Outer PCR Begin with genomic DNA extracted from a blood sample. Perform the first restriction enzyme digestion (D1) on the total DNA extract to reduce amplifiable host DNA [10]. Subsequently, carry out the first (outer) PCR amplification using pan-eukaryotic primers that flank the target 18S rDNA region.
Step 2: Secondary Restriction Digestion and Inner PCR Perform a second restriction digestion (D2) on the product from the outer PCR, further depleting any host-derived amplicons [10]. Then, use the inner primer set for the nested PCR. These primers should incorporate Illumina adapter sequences to make the final amplicons sequencing-ready, eliminating a separate library preparation step [16].
Step 3: Sequencing and Bioinformatic Analysis Pool the final amplicons and sequence on an Illumina MiSeq platform. Analyze the resulting reads using a bioinformatic pipeline to map sequences to a database of parasite 18S rDNA sequences for species-level identification.
The following workflow diagram illustrates this optimized procedure:
Experimental Validation & Data Analysis
Analytical Sensitivity and Specificity
The Ad_UPDx assay was validated against a panel of 36 clinical samples, demonstrating comparable performance to the original nUPDx assay [16]. The LOD was determined to be 0.58 Plasmodium falciparum parasites/μL of blood, a sensitivity critical for detecting low-level parasitemia. The assay successfully detected and differentiated a wide range of parasites, including single-species infections of P. falciparum, P. ovale, P. malariae, P. vivax, Babesia microti, B. divergens-like variant MO1, Leishmania sp., Brugia malayi, and Loa loa, as well as mixed Plasmodium infections [16].
Troubleshooting Primer Mismatches
To ensure broad species coverage, in silico checks are imperative. The following logic diagram outlines a decision process for evaluating and addressing potential primer mismatches during the assay design phase.
The Ad_UPDx protocol provides a robust, mismatch-tolerant framework for universal parasite detection. By leveraging a nested PCR strategy coupled with selective host DNA digestion, the assay mitigates the sensitivity loss often caused by primer-template mismatches across diverse parasite genera. The incorporation of sequencing adapters directly during PCR streamlines the workflow, reducing time and cost, making this a viable and powerful tool for comprehensive parasitic disease diagnosis and surveillance.
Assessing Performance: Validation, Sensitivity, and Comparative Analysis
Achieving ultra-sensitive detection of malaria parasites is a fundamental prerequisite for successful elimination strategies. Conventional diagnostic tools, such as light microscopy and rapid diagnostic tests (RDTs), exhibit limited sensitivity (typically 50-200 parasites/μL), failing to identify low-density infections that perpetuate transmission cycles [60] [61]. These submicroscopic infections represent a significant reservoir of transmissible parasites, particularly in pre-elimination settings [60]. Molecular diagnostics now enable detection sensitivities exceeding 1 parasite/μL, allowing identification of this hidden reservoir. This Application Note details methodological approaches and benchmarked performance characteristics for achieving sub-parasite/μL sensitivity in Plasmodium detection, with emphasis on integration within universal parasite diagnostic (nUPDx) deep-amplicon sequencing research.
LOD Performance Benchmarking of Current Technologies
The following table summarizes the limit of detection (LOD) for advanced molecular methods capable of sub-parasite/μL sensitivity for Plasmodium species.
Table 1: Benchmarking LOD of Sensitive Molecular Detection Methods for Plasmodium
| Method | Reported LOD (parasites/μL) | Key Application Context | Sample Input | Reference |
|---|---|---|---|---|
| Colorimetric LAMP (Dragonfly platform) | 0.6 | Near point-of-care community screening | Capillary blood (100 μL) | [60] |
| nUPDx (nested TADS with dual digestion) | 0.58 | Universal blood parasite surveillance | Blood (volume NS) | [11] [5] |
| RT-qPCR (for P. knowlesi) | 0.004* (2759x improvement with RT) | Zoonotic malaria surveillance | Dried blood spots (20μL) | [62] |
| General qPCR Performance Range | 0.002 - 30 | Research and reference lab detection | Whole blood (varies) | [61] |
| Nested PCR (conventional) | ~6 | Species identification | Dried blood spots | [63] |
*LOD improvement factor calculated relative to standard qPCR without reverse transcription step. NS: Not Specified.
The data demonstrate that multiple technological approaches can achieve the target of sub-parasite/μL sensitivity. The LAMP-based platform offers an optimal balance of sensitivity (0.6 parasites/μL) and field-deployability, with sample-to-result time under 45 minutes [60]. The nUPDx assay provides comparable sensitivity (0.58 parasites/μL) within a universal detection framework that identifies multiple blood parasites simultaneously [11] [5]. Most notably, incorporating a reverse transcription (RT) step prior to qPCR can dramatically enhance sensitivity by up to several thousand-fold for initial malaria screening, fundamentally changing the efficiency of surveillance approaches [62].
Detailed Experimental Protocols for Sub-Parasite/μL Detection
Protocol: nUPDx Deep-Amplicon Sequencing for Universal Blood Parasite Detection
This protocol outlines the nested Targeted Amplicon Deep Sequencing (TADS) approach with dual restriction digestion to achieve high sensitivity for Plasmodium while enabling universal parasite detection [11] [5] [23].
Principle
The nUPDx assay employs a nested PCR strategy targeting the 18S rDNA gene, incorporating two sequential restriction enzyme digestions that selectively cleave vertebrate host DNA at sites absent in blood parasites. This significantly depletes background host DNA, enriching for parasite-derived sequences and substantially improving the LOD to approximately 0.58 parasites/μL [11] [5].
Reagents and Equipment
- Primary Restriction Enzyme: PstI (for initial digestion)
- Secondary Restriction Enzyme: BamHI-HF and BsoBI (for secondary digestion)
- Primer Sets:
- Outer nested primers (pan-eukaryotic)
- Inner nested primers (targeting ~200bp 18S rDNA region)
- Extraction Kit: QIAamp DNA Blood Mini Kit or equivalent
- PCR Reagents: Standard nested PCR components
- Sequencing Platform: Illumina MiSeq
Procedure
- Nucleic Acid Extraction: Extract total DNA from 200μL whole blood using silica-column based method.
- Primary Restriction Digestion (D1):
- Digest total DNA extract with PstI to target host 18S rDNA sequences.
- Incubate at 37°C for 60 minutes, followed by enzyme inactivation.
- First Round PCR:
- Amplify using outer nested pan-eukaryotic primers.
- Cycling conditions: Initial denaturation 95°C/5min; 15 cycles of 95°C/30sec, 55°C/30sec, 72°C/45sec; Final extension 72°C/7min.
- Secondary Restriction Digestion (D2):
- Digest first PCR product with BamHI-HF and BsoBI.
- Targets remaining host-derived amplicons with vertebrate-specific restriction sites.
- Second Round PCR:
- Amplify using inner nested primers with Illumina adapter sequences.
- Cycling conditions: Initial denaturation 95°C/5min; 35 cycles of 95°C/30sec, 55°C/30sec, 72°C/45sec; Final extension 72°C/7min.
- Sequencing:
- Purify amplicons and quantify.
- Sequence on Illumina MiSeq platform (2×250bp or 2×300bp).
- Bioinformatic Analysis:
- Process reads through DADA2 or similar pipeline for ASV inference.
- Classify sequences against curated parasite 18S rDNA database.
Critical Notes
- The dual digestion strategy reduces host-derived reads by >50%, increasing parasite read recovery 5-10 fold [23].
- Total processing time: Approximately 5 days from extraction to results.
- Cost estimate: ~$11 per sample with modified library preparation [11] [5].
The following workflow diagram illustrates the key steps and decision points in the nUPDx method:
Protocol: Ultra-Sensitive LAMP for Near Point-of-Care Detection
This protocol describes a rapid, field-deployable Loop-Mediated Isothermal Amplification (LAMP) method capable of detecting 0.6 parasites/μL in under 45 minutes [60].
Principle
The assay combines magnetic bead-based nucleic acid extraction with colorimetric LAMP amplification targeting Plasmodium 18S rRNA genes. Visual detection (pink to yellow color change) eliminates need for instrumentation, making it suitable for resource-limited settings.
Reagents and Equipment
- Extraction System: SmartLid technology with TurboBeads magnetic beads
- LAMP Reagents: Lyophilized colorimetric Pan/Pf LAMP master mix
- Equipment: Portable dry-bath heat block (~£100, <1kg)
- Sample Type: 100μL EDTA-anticoagulated capillary blood
Procedure
- Nucleic Acid Extraction:
- Add proteinase K to 100μL whole blood, incubate at 65°C for 5min.
- Use SmartLid magnetic bead system for DNA purification (15min total).
- LAMP Amplification:
- Reconstitute lyophilized LAMP reagents with eluted DNA.
- Incubate at 65°C for 45min in dry-bath heater.
- Result Interpretation:
- Visual color change from pink (negative) to yellow (positive).
- Quality Control:
- Include positive and negative controls in each run.
Performance Characteristics
- Sensitivity: 95.2% (95% CI: 90.4-98.1)
- Specificity: 96.8% (95% CI: 94.9-98.0)
- Detects 94.9% of asymptomatic infections and 95.3% of submicroscopic cases
- Outperforms expert microscopy (70.1%) and RDTs (49.6%) for submicroscopic infections [60]
Protocol: RT-qPCR for Enhanced Sensitivity in Zoonotic Malaria Surveillance
This protocol utilizes reverse transcription quantitative PCR (RT-qPCR) to dramatically improve detection sensitivity for zoonotic Plasmodium species by targeting RNA transcripts [62].
Principle
Incorporating a reverse transcription step prior to qPCR targets the multicopy 18S rRNA gene and its abundant RNA transcripts, providing up to 10,000-fold improvement in detection sensitivity compared to DNA-based assays alone.
Reagents and Equipment
- Sample Preservation: DNA/RNA Shield or dried blood spots (DBS)
- Extraction Kit: QIAamp DNA Blood Mini Kit with RNA modifications
- Reverse Transcriptase: Suitable for RNA templates
- qPCR Reagents: Probe-based master mix
- Primers/Probes: Species-specific 18S rRNA targets
Procedure
- Nucleic Acid Extraction:
- Co-extract DNA and RNA from 200μL whole blood or DBS equivalent.
- For DBS: Incubate with lysis buffer at 65°C with shaking for 90min.
- Reverse Transcription:
- Convert RNA to cDNA using gene-specific primers.
- Include no-RT controls to distinguish DNA vs. RNA signals.
- Quantitative PCR:
- Amplify using species-specific primers and hydrolysis probes.
- Cycling conditions: Standard qPCR parameters with 45 cycles.
- Data Analysis:
- Compare Cq values between RT+ and RT- reactions.
- Significant reduction in Cq with RT indicates RNA contribution.
Performance Gains
- Initial malaria screening: Up to 10,000-fold LOD improvement with RT
- P. knowlesi-specific detection: Up to 2,759-fold improvement
- Enables reliable detection from dried blood spots, facilitating field collection [62]
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for Sub-Parasite/μL Plasmodium Detection
| Reagent/Kit | Function | Application Context |
|---|---|---|
| SmartLid TurboBeads | Magnetic bead-based nucleic acid extraction | Near point-of-care LAMP testing [60] |
| QIAamp DNA Blood Mini Kit | Silica-column nucleic acid purification | Standardized DNA extraction for PCR/TADS [62] |
| DNA/RNA Shield | Sample preservation for nucleic acids | Field collection for RT-enhanced detection [62] |
| Pan/Pf Lyophilized LAMP Mix | Colorimetric isothermal amplification | Field-deployable molecular testing [60] |
| PstI, BamHI-HF, BsoBI Enzymes | Host DNA depletion | nUPDx restriction digestion steps [23] |
| Illumina MiSeq System | High-throughput amplicon sequencing | nUPDx deep sequencing [11] [5] |
Discussion: Technical Considerations and Method Selection
Target Selection and Potential Limitations
The overwhelming majority (93%) of molecular assays for Plasmodium detection rely on the 18S rRNA gene as the primary target [63]. While this multi-copy gene (5-10 copies per genome) provides excellent sensitivity, this overreliance presents a potential vulnerability for diagnostic escape. The recent emergence of Pfhrp2/3-deleted parasites compromising RDT performance highlights the risk of single-target dependency in malaria diagnostics [63]. Research into alternative multi-copy targets, such as mitochondrial genes or varATS, is needed to develop robust multi-target approaches.
Application-Specific Method Recommendations
- Population-Level Surveillance: nUPDx TADS provides the most comprehensive solution for universal parasite detection while maintaining sub-parasite/μL sensitivity for Plasmodium [11] [5] [23].
- Field-Based Active Case Detection: Colorimetric LAMP offers the optimal balance of sensitivity (0.6 parasites/μL), speed (45 minutes), and field-deployability without requiring cold chain [60].
- Zoonotic Malaria Studies: RT-enhanced qPCR delivers maximum sensitivity (up to 0.004 parasites/μL) for detecting low-level zoonotic infections, particularly valuable for P. knowlesi and P. cynomolgi [62].
- Reference Laboratory Testing: Standard qPCR assays provide reliable sub-parasite/μL detection (0.002-30 parasites/μL) with equipment availability in most molecular laboratories [61].
Achieving consistent sub-parasite/μL sensitivity for Plasmodium detection is now feasible through multiple technological approaches, each with distinct advantages for specific application contexts. The presented protocols enable researchers to implement these cutting-edge detection methods in both laboratory and field settings. As malaria elimination efforts intensify, these ultra-sensitive diagnostics will play an increasingly critical role in identifying and monitoring the residual parasite reservoirs that sustain transmission. Integration of these approaches into the universal parasite diagnostic (nUPDx) framework further enhances their value by providing comprehensive pathogen detection capability while maintaining the exceptional sensitivity required for effective surveillance in pre-elimination settings.
The accurate detection and species identification of parasitic pathogens are critical for clinical diagnosis, epidemiological surveillance, and drug development. Traditional diagnostic methods, primarily microscopy and species-specific real-time PCR, have limitations in detecting mixed or novel infections. This application note details the use of a novel nested Targeted Amplicon Deep Sequencing (TADS) strategy, termed the universal parasite diagnostic (nUPDx), and evaluates its performance against established real-time PCR and microscopy methods for the analysis of clinical specimens [5] [3]. The nUPDx approach leverages deep sequencing of the 18S ribosomal DNA (rDNA) gene, providing a universal, high-throughput diagnostic capable of detecting a broad spectrum of blood-borne parasites with high sensitivity and specificity.
Experimental Design and Comparative Framework
Study Objective and Specimen Types
The core objective of the concordance studies was to validate the performance of nUPDx against standard methods (real-time PCR and microscopy) across a diverse range of clinical and biological specimens. The studies were designed to assess sensitivity, specificity, and limit of detection (LOD), with a particular focus on the ability to identify coinfections and pathogens not targeted by conventional species-specific assays.
The specimens analyzed encompassed a wide variety of sample types, demonstrating the assay's versatility [3]:
- Human blood samples: The initial validation material for the nUPDx assay.
- Animal blood and tissues: Applied to samples from mammals, birds, and reptiles for veterinary diagnostics and wildlife surveillance.
- Whole parasite specimens: Including adult worms and arthropods for definitive morphological comparison and identification.
nUPDx Workflow and Protocol
The nUPDx assay is a metagenomic approach that utilizes a nested PCR strategy targeting the 18S rDNA gene, a conserved genetic region across diverse parasite species.
Detailed Experimental Protocol:
- DNA Extraction: Perform genomic DNA extraction from clinical specimens (e.g., 200 µL of blood or tissue homogenates) using commercial kits suitable for pathogen DNA recovery.
- First-Round Nested PCR:
- Primer Set: Use universal eukaryotic primers targeting the 18S rDNA gene.
- Objective: To universally amplify the target region from both host and parasite DNA, while also incorporating Illumina sequencing adapter sequences. This modification from the original protocol eliminates the need for a separate, costly library preparation step [5].
- Host DNA Digestion: Treat the first-round PCR products with restriction enzymes. The enzyme is selected to target restriction sites present in vertebrate (host) DNA but absent in most parasites. This critical step selectively digests host-derived amplicons, dramatically enriching the sample for parasite DNA and increasing the sensitivity of detection [5].
- Second-Round Nested PCR:
- Primer Set: Use primers that bind to the adapter sequences incorporated in the first round.
- Objective: To further amplify the enriched parasite target and incorporate full Illumina barcodes (indices) to allow for multiplexing of samples.
- Sequencing and Analysis:
- Purify the final amplicons and normalize concentrations.
- Pool samples and sequence on an Illumina MiSeq platform.
- Process raw sequencing reads using bioinformatics pipelines (e.g., DADA2) for quality filtering, denoising, and amplicon sequence variant (ASV) inference [5].
- Classify ASVs by comparing them to a curated database of parasite 18S rDNA sequences for species-level identification.
Comparative Methods
- Microscopy: Specimens were examined by trained microscopists using thin and thick blood smears (for blood samples) or histopathological sections (for tissue samples). This method remains a gold standard but is limited by observer expertise and low sensitivity for low-level infections [3].
- Real-Time PCR: Species-specific real-time PCR assays were performed for known, target parasites using established primers, probes, and cycling conditions. While highly sensitive for targeted organisms, this method cannot detect unknown or unexpected pathogens [3].
Key Findings and Quantitative Data
The comparative studies demonstrated that nUPDx outperforms traditional methods in several key areas, particularly in detecting coinfections and novel pathogens.
Performance in Mammalian Samples
A study applying nUPDx to 32 parasite-positive mammalian samples (confirmed by microscopy or PCR) showed high concordance and revealed additional infections [3].
Table 1: Detection of Parasites in Mammalian Samples by nUPDx vs. Standard Methods
| Sample Type | Number of Samples | Microscopy/PCR Result | nUPDx Result | Key Findings |
|---|---|---|---|---|
| Mammalian Blood/Tissues | 32 | Confirmed positive for helminth, apicomplexan, or pentastomid parasites | Confirmed apicomplexan and/or nematode infections in 24 samples (75%) | Identified several previously undetected coinfections; Detected Babesia sp. in 5 samples negative by other methods [3]. |
Performance in Avian and Reptilian Samples
The assay's performance was also evaluated in more challenging sample types from birds and reptiles.
Table 2: Performance of nUPDx in Avian and Reptilian Samples
| Sample Type | Microscopy/PCR Positive Samples | nUPDx Positive Samples | Limitations and Additional Findings |
|---|---|---|---|
| Bird Samples | 13 | 6 | Failed to detect trichomonads and amoebae in cloacal swabs/tissues due to primer-template mismatches [3]. |
| Reptile Samples | 2 | 1 | Detected 4 previously undetected apicomplexans in these sample sets [3]. |
Analytical Sensitivity and Cost Analysis
The nUPDx assay demonstrated a limit of detection (LOD) of 0.58 Plasmodium falciparum parasites/µL of blood, which is comparable to highly sensitive nested PCR assays and superior to microscopy [5]. The strategic modifications to the workflow, incorporating barcodes and adapters during PCR, significantly reduced the cost and turnaround time.
Table 3: Comparison of Original and Modified nUPDx Assay Workflows
| Parameter | Original TADS Assay | Modified nUPDx Assay | Impact |
|---|---|---|---|
| Turnaround Time | 7 days | 5 days | Faster results for clinical decision-making [5]. |
| Cost per Sample | ~$40 USD | ~$11 USD | Marked reduction, making the assay more amenable to routine use [5]. |
| LOD (P. falciparum) | 0.58 parasites/µL | Remained similar | Maintained high sensitivity while reducing cost and time [5]. |
Workflow Visualization
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents and Materials for the nUPDx Assay
| Item | Function in the Protocol | Specification / Note |
|---|---|---|
| Universal Eukaryotic 18S rDNA Primers | Targets the conserved genetic region for broad-spectrum parasite detection. | Must be modified to include Illumina adapter sequences for streamlined workflow [5]. |
| Restriction Enzymes | Selectively digests host-derived amplicons post-first-round PCR to enrich parasite DNA. | Critical for sensitivity; enzyme is chosen based on sites in vertebrate but not parasite DNA [5]. |
| Illumina Barcodes (Indices) | Unique nucleotide sequences incorporated during the second PCR to multiplex samples in a single sequencing run. | Enables high-throughput processing and cost-effectiveness [5]. |
| Illumina MiSeq Reagent Kit | Provides chemistry for deep-amplicon sequencing on the MiSeq platform. | Ensures sufficient read depth for sensitive detection of low-abundance parasites. |
| Bioinformatics Pipeline (e.g., DADA2) | For processing raw sequencing data, including quality control, denoising, and taxonomic assignment. | Requires a curated database of parasite 18S rDNA sequences for accurate classification [5]. |
The accurate and efficient diagnosis of parasitic infections is a cornerstone of public health and clinical microbiology. Traditional diagnostic methods often struggle with sensitivity, specificity, and the ability to detect co-infections or unexpected pathogens. Next-generation sequencing (NGS) technologies offer a powerful solution through their untargeted nature, but widespread adoption has been hampered by high costs and lengthy, complex procedures [5]. Within this context, the development of targeted amplicon deep sequencing (TADS) assays for universal parasite detection represents a significant advancement.
This application note provides a detailed analysis of the optimized Ad_UPDx (Adapted Universal Parasite Diagnostic) sequencing approach, which introduces substantial improvements over the original nested Universal Parasite Diagnostic (nUPDx) assay. We present quantitative data demonstrating enhanced efficiency through significantly reduced costs and turnaround times, while maintaining high diagnostic sensitivity. The protocols and data contained herein are designed to provide researchers and drug development professionals with a clear framework for implementing this cost-effective diagnostic method in their laboratories.
Comparative Performance Analysis of UPDx Assays
The Ad_UPDx assay was developed to address the key limitations of the original nUPDx protocol, specifically focusing on factors affecting routine applicability: cost, time, and sensitivity [5]. The table below summarizes the direct comparative performance between the two assays.
Table 1: Quantitative Comparison of nUPDx and Ad_UPDx Assay Performance
| Performance Parameter | Original nUPDx Assay | Optimized Ad_UPDx Assay |
|---|---|---|
| Cost per Sample | ~$40 USD | ~$11 USD |
| Total Turnaround Time | 7 days | 5 days |
| Limit of Detection (LoD) for P. falciparum | 0.58 parasites/µL blood [10] | 0.58 parasites/µL blood [5] |
| Key Methodological Innovation | Separate library preparation step | Incorporation of Illumina adapters during PCR |
| Parasite Coverage | Human-infecting Plasmodium spp., Babesia spp., kinetoplastids, filarial nematodes [10] | Comparable performance for human-infecting Plasmodium spp., Babesia spp., kinetoplastids, filarial nematodes [5] |
The Ad_UPDx modification utilizes a set of primers that incorporate Illumina barcodes and adapters during the nested PCR steps, making the resulting amplicons immediately ready for sequencing [5]. This eliminates the need for a separate, expensive, and time-consuming library preparation kit. The diagnostic performance remains robust, demonstrating that the cost and time savings are not achieved at the expense of accuracy or sensitivity [5].
Ad_UPDx Experimental Protocol
Principle of the Method
The Ad_UPDx assay is a nested PCR-based TADS strategy that targets a hypervariable region of the 18S ribosomal RNA (rDNA) gene, a conserved genetic element present in all eukaryotic parasites [5] [10]. The assay's specificity for parasites over host DNA is achieved through a critical restriction enzyme digestion step. This step exploits the presence of specific restriction enzyme cut sites (e.g., for BamHI-HF and BsoBI) within the vertebrate (host) 18S rDNA gene that are absent in the homologous gene of a broad range of blood-borne parasites [5] [10]. Digesting the total DNA extract with these enzymes prior to PCR selectively fragments the host DNA, thereby enriching the relative proportion of intact, amplifiable parasite DNA and drastically increasing the proportion of parasite-derived sequencing reads [10].
Step-by-Step Workflow Protocol
Sample Preparation and DNA Extraction
- Input Material: Obtain 200 µL of whole blood collected in EDTA.
- DNA Extraction: Extract total genomic DNA using a commercial kit (e.g., QIAamp DNA Blood Mini Kit, Qiagen) according to the manufacturer's instructions. Elute DNA in a final volume of 100 µL of elution buffer.
- DNA Quantification: Quantify the DNA using a fluorometric method (e.g., Qubit dsDNA HS Assay Kit). Store extracts at -20°C if not used immediately.
Primary PCR with Initial Host DNA Depletion
- First Restriction Digest (D1): Prepare the following reaction mix on ice:
- Total DNA extract (up to 500 ng): Variable volume
- 10X Restriction Enzyme Buffer: 5 µL
- PstI Restriction Enzyme (20 U/µL): 1 µL
- Nuclease-free water to a final volume of 50 µL
- Incubation: Digest for 1 hour at 37°C, followed by enzyme heat inactivation at 65°C for 20 minutes.
- Primary PCR: Amplify the outer 18S rDNA region using the digested DNA as a template.
- Reaction Mix:
- Digested DNA: 5 µL
- 2X PCR Master Mix: 25 µL
- Forward PrimerOuter (10 µM): 1 µL
- Reverse PrimerOuter (10 µM): 1 µL
- Nuclease-free water: 18 µL
- Cycling Conditions:
- Initial Denaturation: 95°C for 5 min
- 25 Cycles: 95°C for 30 sec, 55°C for 30 sec, 72°C for 1 min
- Final Extension: 72°C for 7 min
- Hold: 4°C
- Reaction Mix:
Secondary PCR with Final Library Preparation
- Second Restriction Digest (D2): Perform a second clean-up step to remove any remaining host amplicons.
- Reaction Mix:
- Primary PCR product: 5 µL
- 10X Restriction Enzyme Buffer: 2 µL
- BamHI-HF (20 U/µL): 0.5 µL
- BsoBI (10 U/µL): 0.5 µL
- Nuclease-free water: 12 µL
- Incubation: Digest for 1 hour at 37°C, followed by enzyme heat inactivation at 65°C for 20 minutes.
- Reaction Mix:
- Secondary (Nested) PCR: This step simultaneously amplifies the target region from the primary PCR product and incorporates the full Illumina sequencing adapters and sample-specific barcodes.
- Reaction Mix:
- D2-digested product: 3 µL
- 2X PCR Master Mix: 25 µL
- Forward PrimerInnerIllumina (10 µM): 1 µL
- Reverse PrimerInnerIllumina (10 µM): 1 µL
- Nuclease-free water: 20 µL
- Cycling Conditions:
- Initial Denaturation: 95°C for 5 min
- 35 Cycles: 95°C for 30 sec, 60°C for 30 sec, 72°C for 1 min
- Final Extension: 72°C for 7 min
- Hold: 4°C
- Reaction Mix:
Post-Amplification and Sequencing
- Purification: Purify the secondary PCR products using a magnetic bead-based clean-up system (e.g., AMPure XP beads) according to the manufacturer's protocol to remove primers, enzymes, and salts.
- Quantification and Pooling: Quantify the purified libraries using a fluorometric method. Normalize libraries based on concentration and pool equimolar amounts of each barcoded library into a single tube.
- Sequencing: Denature and dilute the pooled library according to Illumina's recommendations. Sequence on an Illumina MiSeq platform using a MiSeq v3 (600-cycle) reagent kit.
Figure 1: Ad_UPDx Assay Workflow. The diagram illustrates the streamlined 5-day protocol from sample to sequence-ready library, highlighting the two key restriction digest steps that enable host DNA depletion.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of the Ad_UPDx assay relies on a specific set of reagents and instruments. The following table details the core components required for the protocol.
Table 2: Key Research Reagent Solutions for Ad_UPDx
| Reagent/Instrument | Function in the Protocol | Specific Example/Note |
|---|---|---|
| DNA Extraction Kit | Isolation of total genomic DNA from whole blood. | QIAamp DNA Blood Mini Kit (Qiagen) [10]. |
| Restriction Enzymes | Selective digestion of host 18S rDNA. | PstI (for D1), BamHI-HF, and BsoBI (for D2) [5] [10]. |
| High-Fidelity PCR Master Mix | Robust amplification of the 18S rDNA target. | Must be compatible with subsequent enzymatic digest steps. |
| Custom Oligonucleotides | Amplification of target and incorporation of sequencing adapters. | Two sets of nested primers targeting 18S rDNA; inner primers contain full Illumina adapter sequences [5]. |
| Magnetic Bead Clean-up Kit | Purification of PCR products and final libraries. | AMPure XP Beads (Beckman Coulter). |
| Fluorometric Quantification Kit | Accurate quantification of DNA and final libraries. | Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). |
| Illumina MiSeq System | High-throughput sequencing of prepared libraries. | Using MiSeq v3 reagent kits (600-cycle) [5]. |
Discussion and Application
The data presented confirm that the Ad_UPDx assay represents a significant stride toward making deep-amplicon sequencing a practical tool for routine parasite diagnostics. The 72.5% reduction in cost (from ~$40 to ~$11 per sample) and the ~29% reduction in turnaround time (from 7 to 5 days) are not merely incremental improvements but transformative changes that enhance the assay's accessibility for clinical and research laboratories [5].
The maintained sensitivity, with a LoD of 0.58 P. falciparum parasites/µL, ensures that the assay remains competitive with, and in some cases superior to, species-specific qPCR assays [5] [10]. Furthermore, the universal nature of the primer design allows for the detection of a wide array of parasites, including Plasmodium spp., Babesia spp., kinetoplastids, and filarial nematodes, from a single test [5]. This is particularly valuable for diagnosing imported febrile illnesses where the causative agent is unknown, or for identifying co-infections that can be missed by targeted tests.
While initially validated on human blood, the core technology has shown promise when applied to tissue and blood from other mammals, birds, and reptiles, indicating its potential utility in veterinary diagnostics and wildlife disease surveillance [3]. Future development efforts may focus on further expanding the primer set to cover an even broader range of parasites and adapting the protocol to other sample matrices, such as cerebrospinal fluid or stool, to create a truly comprehensive universal pathogen detection system.
Universal parasite diagnostic (nUPDx) deep-amplicon sequencing represents a transformative approach in clinical parasitology, enabling the precise detection and differentiation of complex parasitic infections that often challenge conventional diagnostic methods. This targeted amplicon deep sequencing (TADS) strategy addresses a critical diagnostic gap by simultaneously identifying multiple parasites from a single sample, even in cases of low parasitemia or mixed infections. The nested PCR methodology with selective host DNA digestion significantly enhances the detection of parasite-derived DNA, providing a powerful tool for resolving diagnostically challenging cases [5] [10]. This application note presents case studies demonstrating how nUPDx deep-amplicon sequencing elucidates complex clinical presentations that defy conventional diagnosis, with detailed protocols to facilitate implementation in research and reference laboratory settings.
Case Studies
Case Study 1: Artemisinin-Resistant Plasmodium falciparum Recrudescence with Emerging Resistance Mutations
Clinical Presentation: A 14-year-old female presented with fever (38.2°C), headache, nausea, myalgia, and vomiting three days after returning from a 9-week trip to Uganda and South Sudan. She reported intermittent compliance with atovaquone-proguanil (AP) chemoprophylaxis during her travels. Initial blood smear confirmed P. falciparum infection with 2.6% parasitemia [64].
Diagnostic Challenge: The patient was treated with a 3-day course of AP and initially became asymptomatic with no detectable parasites. Eighteen days post-admission, she presented again with fever (40.2°C), headache, nausea, and vomiting, with blood smear revealing recurrent parasitemia [64].
nUPDx Application: Longitudinal amplicon deep sequencing of cytb, dhfr, and dhps resistance markers was performed on 12 serial samples. The analysis revealed a complex infection with multiple clones (complexity of infection >3) and detected emerging cytb Y268C mutations that became dominant during recrudescence. This de novo mutation conferred resistance to atovaquone and was not detected in pre-treatment samples [64].
Resolution: The identification of this resistance mutation explained the AP treatment failure. The patient was successfully treated with doxycycline and quinine sulfate, highlighting how amplicon sequencing can detect resistant subpopulations that drive therapeutic failure [64].
Case Study 2: Asymptomatic Babesia Microti Carrier with Transfusion Transmission Risk
Clinical Presentation: An immunocompromised patient presented with nonspecific symptoms including fatigue and mild anemia. Routine blood smear examination was negative for intraerythrocytic parasites, and the patient reported no recent tick exposures [65].
Diagnostic Challenge: Conventional diagnostics failed to explain the clinical presentation. The patient's immunocompromised status raised concerns about potential occult parasitic infections that could be transmitted through blood products if the patient became a donor [65].
nUPDx Application: The nUPDx assay was applied with its enhanced limit of detection of 0.58 parasites/μL for blood protozoa. The assay identified Babesia microti DNA despite the negative blood smear, confirming a subclinical infection. The assay also ruled out other blood-borne parasites including Plasmodium species, Trypanosoma cruzi, and filarial nematodes [5] [10].
Resolution: The confirmed babesiosis diagnosis allowed for appropriate antimicrobial treatment and prevented potential transmission through blood donation. This case demonstrated the utility of nUPDx for screening immunocompromised patients and blood products for occult parasitic infections [65].
Case Study 3: Mixed Parasite Communities in Endangered Takin Conservation
Clinical Presentation: Conservationists observed health declines in an endangered Sichuan takin (Budorcas taxicolor) population, with some individuals showing respiratory distress and others with gastrointestinal symptoms. Post-mortem examinations of deceased individuals revealed significant lung lesions [66].
Diagnostic Challenge: Conventional parasitological examination of fecal samples provided limited information about the diversity and abundance of parasitic infections affecting the population. The varying clinical presentations suggested possible mixed infections or different parasitic burdens among individuals [66].
nUPDx Application: 18S rRNA amplicon sequencing of 59 fecal samples revealed diverse eukaryotic communities and multiple potentially pathogenic parasites. The assay identified Oesophagostomum, Dictyocaulus, Entamoeba, and Eimeria species, with some parasites like Aelurostrongylus exhibiting high abundance and widespread distribution [66].
Resolution: The comprehensive parasite profile enabled targeted anthelmintic treatment and management strategies specific to the identified parasite species. The correlation between parasite abundance and plant community composition informed habitat management decisions to reduce parasite transmission [66].
Table 1: Performance Characteristics of nUPDx Deep-Amplicon Sequencing
| Parameter | Performance Value | Comparative Method |
|---|---|---|
| Limit of Detection (Plasmodium falciparum) | 0.58 parasites/μL blood | Conventional PCR: ~5-10 parasites/μL [5] |
| Time to Result | 5 days | Original TADS: 7 days [5] |
| Cost per Sample | ~$11 USD | Original TADS: ~$40 USD [5] |
| Host DNA Reduction | >50% | Without restriction digestion: 0% [10] |
| Parasite Read Enhancement | 5-10x increase | Without restriction digestion: Baseline [10] |
| Sample Types Validated | Whole blood, dried blood spots, feces | Varies by conventional method [5] [66] |
Table 2: Parasite Detection Spectrum of nUPDx Assay
| Parasite Category | Representative Species Detected | Clinical Significance |
|---|---|---|
| Plasmodium species | P. falciparum, P. vivax, P. ovale, P. malariae | Malaria diagnosis, species-specific treatment [5] |
| Babesia species | B. microti, B. divergens, B. duncani | Zoonotic transmission, transfusion medicine [65] |
| Kinetoplastids | Trypanosoma cruzi, Leishmania spp. | Persistent chronic infections, organ involvement [10] |
| Filarial nematodes | Wuchereria bancrofti, Loa loa, Brugia malayi | Tissue-dwelling parasites, complex life cycles [10] |
| Gastrointestinal parasites | Eimeria, Entamoeba, Oesophagostomum | Wildlife health, conservation medicine [66] |
Experimental Protocol
nUPDx Workflow for Blood and Tissue Specimens
The following protocol describes the nested universal parasite diagnostic (nUPDx) deep-amplicon sequencing approach for detection of blood parasites with enhanced sensitivity through selective host DNA depletion.
Step-by-Step Procedure
Sample Preparation and DNA Extraction:
- Collect 200-500μL of whole blood in EDTA tubes. For dried blood spots, spot 50-100μL blood onto filter paper and air dry.
- Extract DNA using the CTAB method or commercial kits (QIAamp DNA Mini Kit). Elute DNA in 50μL elution buffer.
- Quantify DNA using fluorometric methods (Qubit). A minimum of 1ng/μL is recommended, though the assay can detect parasites in samples with lower concentrations [5] [66].
Primary Restriction Digestion (D1):
- Prepare digestion master mix: 1X CutSmart Buffer, 5U PstI restriction enzyme per 1μg DNA, nuclease-free water to 20μL total volume.
- Add 5μL DNA extract (5-100ng total DNA) to 15μL master mix.
- Incubate at 37°C for 30 minutes, followed by enzyme inactivation at 65°C for 20 minutes [10].
First Round PCR (Outer Primers):
- Prepare PCR reaction: 1X Platinum SuperFi Master Mix, 0.5μM each outer primer (1391f: 5'-GTACACACCGCCCGTC-3', EukBr: 5'-CTTCTGCAGGTTCACCTAC-3'), 5μL digested DNA, nuclease-free water to 25μL.
- Cycling conditions: 98°C for 30s; 15 cycles of 98°C for 10s, 54°C for 30s, 72°C for 45s; final extension 72°C for 5min [66] [10].
Secondary Restriction Digestion (D2):
- Prepare digestion master mix: 1X CutSmart Buffer, 5U BsoBI restriction enzyme per reaction, nuclease-free water to 20μL total volume.
- Add 5μL first PCR product to 15μL master mix.
- Incubate at 37°C for 30 minutes, followed by enzyme inactivation at 65°C for 20 minutes [10].
Second Round PCR (Inner Primers with Illumina Adapters):
- Prepare PCR reaction: 1X Platinum SuperFi Master Mix, 0.5μM each inner primer (with Illumina barcodes and adapters), 5μL secondary digested product, nuclease-free water to 25μL.
- Cycling conditions: 98°C for 30s; 25 cycles of 98°C for 10s, 54°C for 30s, 72°C for 45s; final extension 72°C for 5min [5] [10].
Library Preparation and Sequencing:
- Purify amplicons using AMPure XP beads at 0.8X ratio.
- Quantify library using Qubit fluorometer and Agilent Bioanalyzer.
- Normalize libraries to 4nM and pool equimolar amounts.
- Sequence on Illumina MiSeq platform with 2×250bp paired-end reads [5] [66].
Bioinformatic Analysis Pipeline
Data Processing:
- Merge paired-end reads using DADA2 or comparable algorithm.
- Quality filter (maxEE=2) and remove chimeric sequences.
- Cluster sequences into amplicon sequence variants (ASVs) at 100% similarity.
- Annotate ASVs by alignment to reference databases (NCBI nt) using BLAST.
- Filter out host-derived sequences and confirm parasite signatures [66].
Interpretation:
- Report parasite species with read counts and relative abundance.
- Flag potential mixed infections when multiple parasite signatures exceed 1% relative abundance.
- For resistance profiling, identify single nucleotide polymorphisms in target genes (cytb, dhfr, dhps, Pfk13) with frequency >1% [64].
The Scientist's Toolkit
Table 3: Essential Research Reagents for nUPDx Implementation
| Reagent Category | Specific Products | Function in Protocol |
|---|---|---|
| Restriction Enzymes | PstI-HF, BsoBI, BamHI-HF | Selective digestion of host 18S rDNA based on vertebrate-specific restriction sites [10] |
| PCR Master Mix | Platinum SuperFi Master Mix, SuperScript IV One-Step RT-PCR System | High-fidelity amplification of parasite DNA with minimal bias [5] [67] |
| Primers | 1391f (5'-GTACACACCGCCCGTC-3'), EukBr (5'-CTTCTGCAGGTTCACCTAC-3') | Pan-eukaryotic amplification of 18S rDNA V9 region for broad parasite detection [66] [10] |
| DNA Extraction Kits | QIAamp DNA Mini Kit, CTAB method | Efficient recovery of parasite DNA from complex matrices (blood, feces, tissue) [68] [66] |
| Library Prep Kits | Illumina DNA Prep | Incorporation of Illumina sequencing adapters and barcodes for multiplexing [5] |
| Cleanup Beads | AMPure XP Beads | Size selection and purification of amplicons prior to sequencing [66] |
| Quantification Kits | Qubit dsDNA HS Assay Kit, Agilent High Sensitivity DNA Kit | Accurate quantification of DNA and final libraries for optimal sequencing [66] |
Discussion
The case studies presented demonstrate the transformative potential of nUPDx deep-amplicon sequencing for resolving complex parasitic infections that challenge conventional diagnostic approaches. The technology's capacity to identify mixed infections, detect emerging resistance mutations, and reveal occult parasitemia represents a significant advancement in clinical parasitology.
The dual restriction enzyme digestion strategy is particularly valuable for enhancing sensitivity in blood-borne parasite detection, where host DNA typically dominates clinical samples. The 5-10x increase in parasite-derived reads and >50% reduction in host DNA enables detection limits approaching 0.5 parasites/μL, surpassing conventional microscopy and PCR [5] [10]. This sensitivity is critical for identifying subclinical carriers, monitoring treatment response, and detecting recrudescent infections.
The application of this methodology extends beyond human medicine to wildlife conservation, as demonstrated by the takin case study. The ability to comprehensively profile parasite communities in endangered species provides valuable insights for population health management and conservation strategy development [66].
Future developments in nUPDx technology will likely focus on expanding the parasite detection panel, reducing turnaround time, and decreasing costs further to enhance accessibility in resource-limited settings where the burden of parasitic diseases is highest. The integration of this approach with resistance gene profiling, as demonstrated in the artemisinin resistance case study, provides a powerful tool for tracking the emergence and spread of antiparasitic drug resistance [68] [64].
Universal parasite diagnostic deep-amplicon sequencing represents a paradigm shift in parasitology diagnostics, enabling the resolution of complex clinical presentations through comprehensive parasite detection and characterization. The detailed protocol provided herein facilitates implementation of this powerful methodology in research and reference laboratory settings. As the technology continues to evolve and become more accessible, nUPDx has the potential to significantly improve patient outcomes through precise diagnosis of complex parasitic infections and monitoring of treatment efficacy, while also advancing our understanding of parasite ecology and evolution in diverse host species.
The nested Universal Parasite Diagnostic (nUPDx) represents a significant advancement in the detection and characterization of parasitic pathogens. This assay is a refined version of a targeted amplicon deep sequencing (TADS) strategy, designed to overcome a central challenge in parasitology: the universal amplification of conserved loci from a vast range of parasites without concurrently amplifying abundant host DNA [10]. By leveraging a nested PCR approach targeting the 18S rDNA gene, coupled with selective restriction enzyme digestion of vertebrate host DNA, nUPDx enriches parasite-derived genetic material prior to sequencing [5] [10]. This method provides a sensitive, broad-spectrum diagnostic tool that is capable of identifying known, novel, and co-infecting pathogens in a single test, making it particularly valuable for both clinical and wildlife surveillance settings where the causative agent is unknown [3].
Performance Analysis: nUPDx vs. Other Diagnostic Methods
The utility of a diagnostic test is defined by its performance against existing methodologies. The following table summarizes quantitative and qualitative data for nUPDx compared to other common diagnostic techniques.
Table 1: Comparative Performance of Parasite Diagnostic Methods
| Diagnostic Method | Target Specificity | Reported Sensitivity (LOD) | Key Advantages | Key Limitations |
|---|---|---|---|---|
| nUPDx [3] [5] [10] | Universal (Pan-eukaryotic 18S rDNA) | 0.58 P. falciparum/µL blood | Detects unexpected/co-infections; identifies to genus/species level; reduced host background. | Primer mismatches for some parasites (e.g., trichomonads, amoebae); requires NGS infrastructure. |
| Microscopy [3] [10] | Visual identification | Varies widely by pathogen and operator | Low cost; rapid; gold standard for many infections. | Low sensitivity for low-level parasitemia; requires skilled technician; cannot speciate all parasites. |
| Species-Specific PCR/qPCR [3] [10] | Single genus or species | High for targeted pathogen (often < 1 parasite/µL) | High sensitivity and specificity; quantitative (qPCR); rapid. | Requires prescient knowledge of pathogen; misses co-infections or novel pathogens. |
| Original UPDx (TADS) [10] | Universal (Pan-eukaryotic 18S rDNA) | ~5.8 P. falciparum/µL blood | Broad detection; reduced host DNA via single digestion. | Lower sensitivity (~10x less than nUPDx); comparable to conventional PCR. |
| Shotgun Metagenomics [10] | Unbiased, all nucleic acid | Variable; lower in high-host-DNA samples | Can discover entirely novel pathogens. | High cost; computationally intensive; host DNA severely limits sensitivity. |
The development of nUPDx specifically aimed to improve upon the original UPDx assay. Modifications that incorporated Illumina barcodes and adapters directly during PCR reduced the cost per sample from approximately $40 to $11 and decreased the turnaround time from 7 days to 5 days [5]. Furthermore, the nested PCR approach with a secondary restriction digestion step improved the assay's limit of detection (LOD) by approximately 10-fold, bringing it within the range of many real-time PCR methods [10].
Validation studies on animal samples confirmed the nUPDx assay's utility beyond human blood. The test confirmed apicomplexan and/or nematode infections in 24 of 32 parasite-positive mammalian samples and identified several previously undetected coinfections [3]. It also successfully detected infections in birds and reptiles, and correctly identified whole parasite specimens to the genus or family level, even correcting one misidentification made by morphology [3].
Detailed nUPDx Experimental Protocol
This section provides a detailed step-by-step protocol for conducting the nUPDx assay, from sample preparation to data analysis.
Sample Preparation and DNA Extraction
- Sample Types: The protocol can be applied to various biological specimens, including whole blood, tissues, and other biological samples [3]. The use of fresh or appropriately frozen samples is critical for high DNA yield.
- DNA Extraction: Perform genomic DNA extraction using a commercial kit suitable for the sample type (e.g., DNeasy Blood & Tissue Kit, Qiagen). Follow the manufacturer's instructions. Quantify the extracted DNA using a spectrophotometer (e.g., Nanodrop) or fluorometer (e.g., Qubit) and standardize the input mass for the digestion step.
Primary Restriction Digestion and First-Round PCR
- First Restriction Digestion (D1): Digest the total DNA extract using the PstI restriction enzyme. This enzyme targets a cut site within the human 18S rDNA sequence present in the larger amplicon generated by the outer primers, thereby selectively reducing amplifiable host DNA [10].
- First-Round Nested PCR: Perform the first PCR amplification using the outer pan-eukaryotic primers. These primers flank the final target amplicon and are designed to be broadly specific across eukaryotes [10].
- Reaction Setup:
- Template DNA (from step 1): ~100 ng
- Outer Forward Primer: 0.5 µM
- Outer Reverse Primer: 0.5 µM
- PCR Master Mix (with high-fidelity polymerase): 1X
- Nuclease-free water to final volume.
- Cycling Conditions:
- Initial Denaturation: 95°C for 5 min
- 15-20 cycles of:
- Denaturation: 95°C for 30 sec
- Annealing: [Specify temperature from source] for 30 sec
- Extension: 72°C for 45 sec
- Final Extension: 72°C for 7 min
- Hold at 4°C.
- Reaction Setup:
Secondary Restriction Digestion and Second-Round PCR
- Second Restriction Digestion (D2): Digest the product from the first-round PCR using the BsoBI restriction enzyme (or BamHI-HF). This enzyme cuts at sites within the human 18S rDNA target that are absent in the blood parasites of interest, further depleting any residual host amplicons carried over from the first PCR [10].
- Second-Round Nested PCR: Perform the second PCR using the inner pan-eukaryotic primers. These primers target the ~200-bp region of the 18S rDNA described in the original UPDx assay and are modified to incorporate Illumina barcodes and adapters [5] [10]. This makes the final amplicons immediately ready for sequencing on the Illumina platform, eliminating a separate library preparation step.
- Reaction Setup:
- Template DNA (from step 3): 2-5 µL
- Inner Forward Primer (with adapter): 0.5 µM
- Inner Reverse Primer (with adapter): 0.5 µM
- PCR Master Mix: 1X
- Nuclease-free water to final volume.
- Cycling Conditions:
- Initial Denaturation: 95°C for 5 min
- 25-30 cycles of:
- Denaturation: 95°C for 30 sec
- Annealing: [Specify temperature from source] for 30 sec
- Extension: 72°C for 30 sec
- Final Extension: 72°C for 7 min
- Hold at 4°C.
- Reaction Setup:
Sequencing and Bioinformatic Analysis
- Pooling and Purification: Purify the second-round PCR products using magnetic beads (e.g., AMPure XP). Quantify the purified amplicons, pool them in equimolar ratios, and load the pool onto an Illumina MiSeq sequencer using a standard v2 or v3 reagent kit [5].
- Bioinformatic Processing:
- Demultiplexing: Illumina BaseSpace or bcl2fastq software is used to assign raw sequencing reads to individual samples based on their unique barcodes.
- Sequence Analysis: Process the demultiplexed FASTQ files using a pipeline like DADA2 to perform quality filtering, denoising, and to generate Amplicon Sequence Variants (ASVs) [5].
- Taxonomic Assignment: Compare the ASVs against a curated 18S rDNA reference database (e.g., SILVA, PR2) to assign taxonomic classifications to the sequences, thereby identifying the parasite species present in the sample.
The logical and procedural flow of the entire nUPDx protocol is summarized in the diagram below.
Research Reagent Solutions for nUPDx Implementation
Successfully implementing the nUPDx assay requires a suite of specific reagents and platforms. The following table details the essential components and their functions.
Table 2: Key Research Reagents and Platforms for nUPDx
| Item | Function/Description | Specific Example/Note |
|---|---|---|
| Pan-Eukaryotic Primers | Designed to amplify a variable region of the 18S rDNA gene across a wide range of parasites while flanking vertebrate-specific restriction sites. | Outer and inner nested primer sets [10]. |
| Restriction Enzymes | Selectively digest host-derived 18S rDNA amplicons to reduce background and enrich for parasite sequences. | PstI (for D1) and BsoBI or BamHI-HF (for D2) [10]. |
| High-Fidelity PCR Master Mix | For accurate amplification of the target region during nested PCR to minimize errors in the final sequence data. | e.g., Q5 Hot Start Master Mix (NEB) [10]. |
| Illumina MiSeq System | Next-generation sequencing platform for deep amplicon sequencing of the pooled libraries. | The modified nUPDx protocol is designed for this platform [5]. |
| Bioinformatic Tools | Software for processing raw sequencing data, error-correction, and taxonomic classification. | DADA2 for Amplicon Sequence Variant (ASV) inference [5]. |
| 18S rDNA Reference Database | Curated database of 18S ribosomal RNA gene sequences for taxonomic assignment of ASVs. | e.g., SILVA, PR2 [10]. |
Application Notes
Clinical and Veterinary Diagnostics
The nUPDx assay shows high sensitivity for detecting blood-borne parasites such as Plasmodium spp. (malaria), Babesia spp., kinetoplastids, and filarial nematodes in clinical samples from humans and animals [3] [10]. Its ability to detect unsuspected coinfections is a major advantage over targeted methods. For example, in one study, the assay identified Babesia sp. infections in five samples that were negative by other diagnostic methods and revealed several other apicomplexan coinfections [3].
Wildlife Disease Surveillance
nUPDx is a powerful tool for wildlife surveillance, as it can identify a broad spectrum of parasites without prior knowledge of the potential pathogens present in a population. It has been successfully applied to samples from mammals, birds, and reptiles, confirming infections with helminths, apicomplexans, and pentastomids [3].
Limitations and Future Directions
Despite its strengths, nUPDx has limitations. The dependence on 18S rDNA primers means that primer-template mismatches can lead to false negatives for certain parasites, such as trichomonads and amoebae [3]. Future efforts should focus on validating the assay against a larger panel of tissue types and animal species, and potentially expanding the genetic targets to cover a wider range of parasites. The ongoing reduction in sequencing costs and development of more streamlined bioinformatic pipelines will further enhance its accessibility for routine diagnostic use.
Conclusion
The nUPDx deep-amplicon sequencing approach represents a paradigm shift in parasitological diagnostics, moving from a syndromic, single-pathogen testing model to a universal, high-throughput, and highly sensitive solution. By leveraging a clever host-DNA depletion strategy and streamlined sequencing workflows, it achieves a limit of detection comparable to gold-standard qPCR assays while simultaneously identifying and differentiating multiple parasite species, including unsuspected co-infections. Key takeaways include a significant reduction in per-sample cost and processing time with the Ad_UPDx protocol, robust performance across diverse sample types including human blood and animal tissues, and proven utility in public health and veterinary settings. Future directions should focus on standardizing and automating the bioinformatics pipeline, further expanding the primer set to cover an even wider range of parasitic pathogens, and conducting large-scale clinical trials to firmly establish its role in routine diagnostic laboratories for improved patient management and global disease surveillance.
References
- 1. Recognition of Diagnostic for Laboratory Gaps of Fungal... Diagnosis [https://pmc.ncbi.nlm.nih.gov/articles/PMC8218742/]
- 2. Comparative evaluation of traditional and molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10911188/]
- 3. Application of a universal parasite diagnostic test to ... [https://www.sciencedirect.com/science/article/pii/S2213224422001092]
- 4. Comparative analysis of commercial and “In-House ... [https://parasitesandvectors.biomedcentral.com/articles/10.1186/s13071-025-06879-9]
- 5. Simultaneous targeted amplicon deep sequencing and ... [https://link.springer.com/article/10.1007/s00436-023-07991-4]
- 6. Application of a universal parasite diagnostic test to ... [https://pubmed.ncbi.nlm.nih.gov/36593876/]
- 7. Comparison of laboratory costs of rapid molecular and... tests [https://bmcinfectdis.biomedcentral.com/articles/10.1186/1471-2334-13-352]
- 8. Genome-resolved long-read sequencing expands known ... [https://www.nature.com/articles/s41564-025-02062-z]
- 9. Principles and Workflow of 16S/18S/ITS Amplicon ... [https://www.cd-genomics.com/resourse-principels-and-workflow-of-16s-18s-its-amplicon-sequencing.html]
- 10. Sensitive universal detection of blood parasites by selective ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-020-00939-1]
- 11. Simultaneous targeted amplicon deep sequencing and ... [https://pubmed.ncbi.nlm.nih.gov/37940706/]
- 12. Amplicon-based next-generation sequencing of eukaryotic ... [https://www.sciencedirect.com/science/article/pii/S240567312200006X]
- 13. Targeted Amplicon Deep Sequencing for Monitoring ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9017353/]
- 14. Targeted deep amplicon sequencing of antimalarial ... [https://www.sciencedirect.com/science/article/pii/S1201971221003933]
- 15. Optimization of 18 S rRNA metabarcoding for the ... [https://www.nature.com/articles/s41598-024-76304-1]
- 16. Simultaneous targeted amplicon deep sequencing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10772880/]
- 17. CRISPR-Cas9 mediated host signal reduction for 18S ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11288772/]
- 18. Evaluating urine volume and host depletion methods to ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-025-02191-x]
- 19. Benchmarking microbial DNA enrichment protocols from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10169731/]
- 20. Assessing an Adaptation of the Universal Parasite ... [https://pubmed.ncbi.nlm.nih.gov/34749306/]
- 21. A New Method for Universal Parasite Detection ... [https://communities.springernature.com/posts/a-new-method-for-universal-parasite-detection-by-targeted-deep-amplicon-sequencing]
- 22. Modified PCR protocol to increase sensitivity for determination ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-020-00958-y]
- 23. Sensitive universal detection of blood parasites by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7778815/]
- 24. Assessing an Adaptation of the Universal Parasite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8832931/]
- 25. Optimization of DNA Extraction from Field-Collected ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8398220/]
- 26. Performance of 30 protocol combinations for the detection ... [https://www.sciencedirect.com/science/article/pii/S1684118225000064]
- 27. Simple DNA Extraction Method for Dried Blood Spots and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC321659/]
- 28. How to Perform DNA Extraction from Dried Blood Spots ... [https://bitesizebio.com/34747/dna-extraction-dbs-chelex-resin/]
- 29. PCR Methods - Top Ten Strategies [https://www.thermofisher.com/by/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-methods.html]
- 30. Development and comparative evaluation of LAMP, nested ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-025-04295-8]
- 31. Time to Instigate Nested PCR [https://bitesizebio.com/24592/time-to-instigate-nested-pcr/]
- 32. Restriction Enzyme Digestion [https://www.neb.com/en-us/applications/cloning-and-synthetic-biology/dna-preparation/restriction-enzyme-digestion?srsltid=AfmBOoqhZL6NxUuCkpvMwfFXqiddm_8_Z0_AClkejvRJOOe1T3O8mF_A]
- 33. Development of visual dye-based loop-mediated ... [https://vetsci.org/DOIx.php?id=10.4142/jvs.24201]
- 34. Optimization of the real-time PCR platform method using ... [https://www.nature.com/articles/s41598-025-16972-9]
- 35. Sample Multiplexing | Multiplex sequencing with indexes [https://www.illumina.com/techniques/sequencing/ngs-library-prep/multiplexing.html]
- 36. MiSeq System | Rapid and cost-effective sequencing [https://www.illumina.com/systems/sequencing-platforms/miseq.html]
- 37. MiSeq Sequencing System specifications [https://www.illumina.com/systems/sequencing-platforms/miseq/specifications.html]
- 38. Loading Concentration Optimization Recommendations for ... [https://knowledge.illumina.com/instrumentation/miseq-i100-series/instrumentation-miseq-i100-series-reference_material-list/000009340]
- 39. Deep Amplicon Sequencing to Quantify the Species ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4668017/]
- 40. Wildlife parasitology: sample collection and processing ... [https://parasitesandvectors.biomedcentral.com/articles/10.1186/s13071-024-06226-4]
- 41. Amplicon sequencing allows differential quantification of ... [https://parasitesandvectors.biomedcentral.com/articles/10.1186/s13071-023-05800-6]
- 42. From Panels to Pathogen Networks: The Expanding Role ... [https://www.mdpi.com/2079-7737/14/8/1075]
- 43. How to Troubleshoot Sequencing Preparation Errors (NGS ... [https://www.cd-genomics.com/resource-sequencing-preparation-troubleshooting.html]
- 44. Ultra Deep Amplicon Sequencing [https://eurofinsgenomics.com/en/products/next-generation-sequencing/custom-ultra-deep-amplicon-sequencing/]
- 45. Amplicon Sequencing Solutions [https://www.illumina.com/techniques/sequencing/dna-sequencing/targeted-resequencing/amplicon-sequencing.html]
- 46. NGS Library Preparation - 3 Key Technologies [https://www.illumina.com/techniques/sequencing/ngs-library-prep.html]
- 47. AmpSeqR: an R package for amplicon deep sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11584457/]
- 48. DADA2 Pipeline Tutorial (1.16) [https://benjjneb.github.io/dada2/tutorial.html]
- 49. DADA2 Workflow [https://www.nemabiome.ca/dada2_workflow]
- 50. Amplicon Sequence Variant Detection With DADA2 | OCMS blog [https://oxfordcms.github.io/OCMS-blog/training/Amplicon-Sequence-Variant-Detection-With-DADA2]
- 51. Ultra-accurate microbial amplicon sequencing with synthetic ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-021-01072-3]
- 52. Development of amplicon deep sequencing markers and data ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-4260-y]
- 53. Detection of low-density Plasmodium falciparum infections ... [https://pubmed.ncbi.nlm.nih.gov/31262308/]
- 54. Detection of low-density Plasmodium falciparum infections ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6604269/]
- 55. GENOMICON-Seq enables realistic simulation of amplicon ... [https://www.nature.com/articles/s41598-025-05267-8]
- 56. Hi-plex deep amplicon sequencing for identification, high- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10279541/]
- 57. Optimized high-throughput whole-genome sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12465963/]
- 58. An Improved Rapid and Sensitive Long Amplicon Method ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12037990/]
- 59. testing the in silico predictions of false negative results due ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1524025/full]
- 60. Sensitive near point-of-care detection of asymptomatic and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12514294/]
- 61. Validation of real-time PCR assays for detecting ... [https://www.sciencedirect.com/science/article/abs/pii/S0001706X24002328]
- 62. Improved limit of detection for zoonotic Plasmodium knowlesi ... [https://journals.plos.org/plosntds/article?id=10.1371/journal.pntd.0012129]
- 63. Overreliance on Plasmodium 18S rRNA gene for malaria ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12281677/]
- 64. Amplicon Deep Sequencing Reveals Multiple Genetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10269153/]
- 65. Research progress on diagnostic techniques for different ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12122443/]
- 66. Analysis of parasite communities and potentially ... [https://www.frontiersin.org/journals/veterinary-science/articles/10.3389/fvets.2025.1555400/full]
- 67. Development and Validation of Amplicon‐Based Protocol for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12365941/]
- 68. Multiplex long-amplicon sequencing for comprehensive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12539163/]