Shape Space and Classification in Morphometrics: From Mathematical Foundations to Biomedical Applications
This article provides a comprehensive exploration of shape space theory and classification methodologies within geometric morphometrics, tailored for researchers and drug development professionals.
Shape Space and Classification in Morphometrics: From Mathematical Foundations to Biomedical Applications
Abstract
This article provides a comprehensive exploration of shape space theory and classification methodologies within geometric morphometrics, tailored for researchers and drug development professionals. It covers the foundational mathematical principles, including key shape space models like Kendall's shape space and differential coordinates. The scope extends to practical applications in drug discovery and clinical assessment, detailing both alignment-based and alignment-free methods. It also addresses critical challenges such as measurement error, data pooling, and the 'out-of-sample' problem, offering optimization strategies and validation protocols. Finally, the article evaluates the performance of different classification techniques and discusses emerging computational trends, synthesizing key takeaways for biomedical research.
The Geometry of Form: Unpacking the Core Principles of Shape Space
The concept of a shape space provides a formal mathematical framework for quantifying and comparing forms in nature, technology, and science. In essence, a shape space is a mathematical construct in which each point represents a distinct shape, and distances between points correspond to quantitative measures of shape dissimilarity [1]. This conceptualization has become fundamental to numerous disciplines, from evolutionary biology and paleontology to pharmaceutical development and materials science. The study of shape space enables researchers to move beyond qualitative descriptions to rigorous statistical analyses of form, variation, and transformation.
The importance of shape space analysis stems from the critical role that form plays in determining function across biological and physical systems. In molecular biology, shape complementarity governs interactions between drugs and their protein targets, antibodies and antigens, and enzymes and their substrates [2]. In evolutionary biology, shape changes in fossil lineages provide evidence for evolutionary processes and environmental adaptations [3]. The quantitative framework of shape space allows researchers to precisely characterize these relationships, test hypotheses about factors affecting form, and visualize complex morphological patterns.
Mathematical Foundations of Shape
Topological Concepts of Shape
Topology, often described as "rubber-sheet geometry," provides the most fundamental mathematical perspective on shape by focusing on properties that remain invariant under continuous deformations such as stretching, bending, and twisting [4]. Unlike classical geometry, which concerns itself with precise distances and angles, topology considers two objects to be equivalent if one can be transformed into the other without tearing or gluing. A circle is thus topologically equivalent to an ellipse or square, while a sphere is equivalent to a cube but not to a torus (doughnut shape) [4].
The mathematical formalization of these concepts occurs through topological spaces, which define the minimal structure needed to discuss continuity and connectedness [5]. A topological space consists of a set of points along with a collection of open sets that satisfy specific axioms governing unions and intersections. This abstract framework enables the definition of key topological properties including:
- Connectedness: Whether a space can be divided into disjoint open sets
- Compactness: A generalization of closed and bounded sets
- Homeomorphism: The fundamental equivalence relation in topology, where a continuous deformation with a continuous inverse exists between two spaces
These topological concepts provide the foundational "glue" for constructing more structured shape spaces, as they define the most basic level of shape equivalence and transformation.
Geometric Morphometrics and Shape Spaces
While topology provides the basic language of shape transformation, geometric morphometrics operationalizes shape analysis for practical scientific applications. Geometric morphometrics defines shape as "all the geometric information that remains when location, scale, and rotational effects are filtered out from an object" [1]. This definition leads directly to the construction of explicit shape spaces with measurable distances between shapes.
The most common approach to constructing such shape spaces uses Procrustes superimposition [1]. This method involves:
- Translation: Centering configurations at the origin
- Scaling: Normalizing to unit centroid size
- Rotation: Aligning configurations to minimize distances between corresponding points
The resulting Procrustes shape coordinates reside in a curved, non-Euclidean space. For statistical analysis, shapes are typically projected into a tangent space that approximates this curved shape space near a reference configuration, enabling the application of conventional multivariate statistics [1].
Table 1: Key Mathematical Spaces for Shape Analysis
| Space Type | Key Properties | Primary Applications |
|---|---|---|
| Topological Space | Defines continuity and connectedness; no metric structure | Qualitative shape classification; fundamental shape equivalence |
| Shape Space | Curved manifold; Procrustes distance metric | Biological morphometrics; comparative anatomy |
| Tangent Space | Euclidean approximation to shape space | Multivariate statistical analysis |
| Form Space | Incorporates size and shape together | Allometric studies; growth analysis |
Methodological Frameworks for Shape Analysis
Landmark-Based Approaches
Landmark-based methods form the cornerstone of modern geometric morphometrics. This approach relies on the precise identification of anatomically homologous points across specimens, classified into three distinct types [1]:
- Type I landmarks: Defined by local tissue geometry (e.g., intersection of three sutures)
- Type II landmarks: Points of maximum or minimum curvature (e.g., tip of a structure)
- Type III landmarks: Extremal points defined by maximal distance from other landmarks
The configuration of landmarks for each specimen is recorded as a matrix of coordinates, which undergoes Procrustes superimposition to extract shape variables [1]. The power of this approach lies in its ability to preserve the geometric relationships among landmarks throughout analysis, enabling sophisticated visualization of shape change through deformation grids and vector diagrams.
Landmark-based methods face limitations when studying structures lacking numerous homologous points or when comparing highly disparate forms. These challenges have led to the development of complementary approaches using semilandmarks, which capture information along curves and surfaces [1].
Landmark-Free Methods
Recent computational advances have enabled landmark-free approaches that capture shape variation without requiring manually identified homologous points. These methods are particularly valuable for analyzing large datasets or structures with few clear landmarks [6].
One prominent landmark-free method is Deterministic Atlas Analysis (DAA), implemented through Large Deformation Diffeomorphic Metric Mapping (LDDMM) [6]. This approach:
- Generates a consensus "atlas" shape through iterative alignment of all specimens
- Calculates deformation fields mapping the atlas to each specimen
- Uses momentum vectors at control points to quantify shape variation
The DAA framework automatically distributes control points throughout the shape, with density controlled by a kernel width parameter [6]. Smaller kernel values produce more control points and capture finer-scale shape details. This method has demonstrated particular utility in large-scale evolutionary studies encompassing highly divergent forms where homologous landmarks become scarce.
Table 2: Comparison of Shape Analysis Methodologies
| Method | Data Type | Key Advantages | Limitations |
|---|---|---|---|
| Traditional Landmarks | Type I-III landmarks | Clear biological homology; well-established statistics | Time-consuming; limited coverage of surfaces |
| Semilandmarks | Points along curves/surfaces | Captures outline and surface geometry | Requires sliding algorithms; arbitrary spacing |
| Outline Analysis | Mathematical functions fitted to outlines | Comprehensive boundary capture; no landmarks needed | Disregards internal homology; sensitive to noise |
| DAA/LDDMM | Dense surface meshes | Automated; comprehensive coverage; no landmarks | Complex implementation; computationally intensive |
Applications Across Scientific Disciplines
Molecular Shape in Drug Discovery
In pharmaceutical research, molecular shape similarity serves as a powerful principle for identifying potential drug candidates, based on the concept that structurally similar molecules often share similar biological properties [7]. Shape-based virtual screening compares the three-dimensional geometry of a query molecule with large databases of compounds to identify those with complementary shapes to target proteins [7] [2].
Multiple computational approaches have been developed to quantify molecular shape similarity:
- Atom-Based Methods: Ultrafast Shape Recognition (USR) and related techniques describe molecular shape using distributions of atomic distances from reference points [7]
- Gaussian Models: Represent atoms as overlapping Gaussian functions to calculate molecular volumes and overlap scores [8]
- Alignment-Based Methods: Optimize the spatial overlap between molecules through rotational and translational adjustments [7]
These methods enable scaffold hopping—identifying compounds with different molecular frameworks but similar overall shapes that may interact with the same biological target [7]. The Tanimoto Similarity Index provides a standardized measure of shape overlap, ranging from 0 (no overlap) to 1 (identical shapes) [8].
Diagram 1: Molecular Shape Similarity Screening Workflow. This flowchart illustrates the computational pipeline for shape-based virtual screening of compound databases.
Biological Morphometrics and Evolution
In evolutionary biology, shape space analysis has revolutionized the study of phenotypic evolution by enabling precise quantification of morphological change [9] [3]. Allometry—the study of size-related shape changes—has been particularly advanced through geometric morphometric frameworks [9]. Two primary schools of thought have emerged:
- Gould-Mosimann School: Defines allometry as the covariation between shape and size, typically analyzed through multivariate regression of shape variables on size measures [9]
- Huxley-Jolicoeur School: Characterizes allometry as covariation among morphological features all containing size information, represented by the first principal component of form space [9]
These approaches have been applied across different biological levels:
- Ontogenetic allometry: Shape changes during growth within a species
- Static allometry: Shape variation among adults of the same species
- Evolutionary allometry: Shape differences across related species or higher taxa
Geometric morphometric analyses have revealed conserved patterns of morphological integration, evolutionary rates varying across structures, and the influence of developmental constraints on evolutionary trajectories [9] [3].
Experimental Protocols and Research Tools
Detailed Methodological Protocols
Protocol 1: Procrustes-Based Geometric Morphometrics
This established protocol for landmark-based shape analysis involves sequential steps to isolate biological shape variation from other sources of geometric differences [1]:
- Landmark Digitization
- Identify and record Type I, II, and III landmarks across all specimens
- Ensure landmarks represent biologically homologous points
- For 3D data, use micro-CT scanners or laser scanners for coordinate acquisition
Procrustes Superimposition
- Translate configurations to a common origin (centroid)
- Scale configurations to unit centroid size
- Rotate configurations to minimize the sum of squared distances between corresponding landmarks
Statistical Analysis in Tangent Space
- Project Procrustes coordinates into a Euclidean tangent space
- Apply multivariate statistical methods (PCA, MANOVA, regression)
- Visualize shape changes along statistical axes using deformation grids
Validation and Visualization
- Assess measurement error through replicate measurements
- Visualize mean shapes and shape changes using thin-plate spline deformations
- Map statistical results back to anatomical space for biological interpretation
Protocol 2: Landmark-Free Shape Analysis Using DAA
This emerging protocol for automated shape analysis is particularly suitable for large datasets and structures lacking clear landmarks [6]:
Data Standardization and Preprocessing
- Convert all specimens to watertight, closed meshes using Poisson surface reconstruction
- Ensure consistent mesh quality and resolution across specimens
- Apply consistent mesh decimation if needed for computational efficiency
Atlas Generation
- Select an initial template specimen representative of morphological diversity
- Iteratively compute a consensus atlas shape minimizing total deformation energy
- Generate control points guided by kernel width parameter (typically 10-40mm)
Shape Registration and Comparison
- Compute diffeomorphic deformations mapping atlas to each specimen
- Calculate momentum vectors at control points quantifying deformation magnitude and direction
- Apply kernel Principal Component Analysis (kPCA) to explore shape variation
Macroevolutionary Analysis
- Compare shape distances with phylogenetic distances
- Calculate morphological disparity across groups
- Estimate evolutionary rates in shape space
Diagram 2: Comparative Workflows for Shape Analysis Methodologies. This diagram contrasts the key stages in landmark-based and landmark-free approaches to shape space analysis.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Tools for Shape Space Analysis
| Tool/Category | Specific Examples | Function/Purpose |
|---|---|---|
| Imaging Modalities | Micro-CT scanners, laser surface scanners, MRI | Generate 3D digital representations of specimens |
| Landmarking Software | tpsDig2, MorphoJ, EVAN Toolbox | Digitize landmarks and perform basic shape analysis |
| Shape Analysis Platforms | R (geomorph package), PAST, Deformetrica | Comprehensive statistical analysis of shape data |
| Molecular Shape Tools | ROCS, USR-VS, OptiPharm | Calculate molecular shape similarity for drug discovery |
| Visualization Software | MeshLab, Landmark Editor, Paraview | Visualize 3D shapes and shape transformations |
The mathematical formalization of shape space has transformed how researchers across diverse fields quantify, compare, and analyze form. From the abstract foundations of topology to the practical applications in drug discovery and evolutionary biology, shape space concepts provide a unified framework for understanding morphological variation. The continuing development of both landmark-based and landmark-free methods ensures that shape analysis can adapt to increasingly large and complex datasets while maintaining biological relevance.
As shape space methodologies evolve, several frontiers appear particularly promising: the integration of developmental dynamics into shape models, the reconciliation of discrete character data with continuous shape variables, and the application of machine learning to identify biologically meaningful shape features automatically. These advances will further solidify shape space as an essential conceptual and analytical framework throughout the scientific disciplines concerned with form and function.
Shape spaces provide a foundational mathematical framework for analyzing and comparing biological forms in morphometrics research. A shape space is a mathematical construct where each point corresponds to a distinct shape, and distances between points quantify shape dissimilarity [10]. The core definition of shape in this context encompasses all geometric features of an object except for its size, position, and orientation [10]. This conceptual separation allows researchers to focus specifically on morphological variation without confounding factors from placement or scale. The development of rigorous shape space theories has provided morphometrics with a firm mathematical foundation for statistical operations such as estimating average shapes and characterizing shape variation within and between populations—operations that are fundamental to biological applications across evolutionary biology, anthropology, and biomedical sciences [10].
The complexity of shape spaces stems from their inherent curvature and multidimensionality, particularly for configurations with more than three landmarks [10]. For biological shapes represented by landmark configurations, the dimensionality of Kendall's shape space can be calculated as 2k-4 for 2D data (where k is the number of landmarks) and 3k-7 for 3D data [10]. This reduction from the original coordinate representation accounts for the non-shape components: in 2D, one dimension is removed for size, two for translation, and one for rotation, while in 3D, one dimension is removed for size, three for translation, and three for rotation [10]. Understanding these foundational concepts is crucial for researchers applying geometric morphometrics to drug development, where precise quantification of morphological changes can reveal treatment effects, toxicity responses, or structural modifications at cellular or organismal levels.
Kendall's Shape Space
Kendall's shape space, named after David G. Kendall, represents one of the most established mathematical frameworks for shape analysis in morphometrics. This approach defines shape as a property that remains after filtering out effects of translation, rotation, and scaling [11]. Mathematically, a Kendall shape space for k landmarks in m dimensions is denoted as Σₘᵏ = Sₘᵏ/SO(m), where Sₘᵏ represents the pre-shape space consisting of centered and normalized configurations (Sₘᵏ := {X ∈ ℝᵐ ˣ ᵏ ‖ ∑ᵢ Xᵢ = 0, ‖X‖ꜰ = 1}), and SO(m) represents the special orthogonal group (rotation matrices) whose action on the pre-shape space is quotiented out [12]. The pre-shape space itself can be identified with a hypersphere 𝕊⁽ᵏ⁻¹⁾ᵐ⁻¹ through a transformation ψ(X) = HX/‖HX‖, where H is a matrix that centers the configuration [12].
Procrustes Distance and Superimposition
The fundamental metric in Kendall's shape space is the Procrustes distance, which quantifies shape difference through a rigorous superimposition process [10]. The procedure for comparing two landmark configurations involves three sequential steps:
- Size normalization: Both configurations are scaled to unit centroid size, where centroid size is defined as the square root of the sum of squared distances of each landmark from the centroid [10].
- Translation: The scaled configurations are shifted so their centroids coincide at the origin [10].
- Rotation: One configuration is rotated around the common centroid to minimize the sum of squared distances between corresponding landmarks [10].
This process exists in two variants: partial Procrustes superimposition (both configurations scaled to unit size) and full Procrustes superimposition (only the target is fixed at unit size while the other is optimally scaled) [10]. The full Procrustes distance represents the minimum Euclidean distance between corresponding landmarks after optimal superimposition [10].
Table 1: Dimensionality of Kendall's Shape Space for Different Landmark Configurations
| Landmarks | Data Type | Original Coordinates | Non-shape Dimensions | Shape Dimensions |
|---|---|---|---|---|
| 3 | 2D | 6 | 4 | 2 |
| 4 | 2D | 8 | 4 | 4 |
| 5 | 2D | 10 | 4 | 6 |
| k | 2D | 2k | 4 | 2k-4 |
| k | 3D | 3k | 7 | 3k-7 |
Visualizing Kendall's Shape Space for Triangles
The simplest non-trivial Kendall shape space is for triangles in 2D, which forms a spherical surface known as the shape sphere [10]. This provides an intuitive model for understanding properties of shape spaces in general. On this sphere, each point represents a distinct triangle shape, with antipodal points representing reflected triangles [10]. Great circles on this sphere correspond to repeated applications of specific shape changes, helping visualize why shape spaces are curved, closed surfaces [10]. For biological datasets with more landmarks, the shape spaces become higher-dimensional, but the spherical nature persists in abstract form, with the curvature having implications for statistical analysis.
Point Distribution Model
The Point Distribution Model (PDM) represents a Euclidean approximation to Kendall's shape space that enables multivariate statistical analysis [11]. This approach linearizes the curved shape space by working in a tangent space projected from a mean shape, creating a vector space where standard multivariate statistical techniques can be directly applied [11]. The PDM is constructed through Procrustes alignment of all specimens to a common reference shape, effectively reducing rotational and translational effects while preserving shape variability in a linear space [11].
Mathematical Foundation and Construction
The Point Distribution Model operates by projecting shapes from the curved Kendall shape space onto a Euclidean tangent space at a specific point, typically the mean shape or a reference shape [11]. The projection is mathematically valid when shape variation is sufficiently small, which empirical analyses suggest is satisfactory to excellent for most biological datasets [10]. The construction process involves:
- Procrustes Superimposition: All specimen configurations are aligned to a reference shape using Generalized Procrustes Analysis (GPA)
- Mean Shape Calculation: The average of aligned shapes is computed
- Tangent Space Projection: Shapes are projected to the Euclidean space tangent to the shape space at the mean shape
- Covariance Estimation: The covariance matrix of the tangent coordinates is computed
- Principal Components Analysis: Eigen decomposition of the covariance matrix identifies major modes of shape variation
Table 2: Comparison of Shape Representation Models
| Model | Mathematical Structure | Invariance | Statistical Framework |
|---|---|---|---|
| Kendall's Shape Space | Riemannian manifold | Rotation, Translation, Scale | Geometric statistics on manifolds |
| Point Distribution Model | Euclidean tangent space | Rotation, Translation (via alignment) | Standard multivariate statistics |
| Differential Coordinates | Lie group structure | Translation | Riemannian geometry on Lie groups |
| Fundamental Coordinates | Lie group structure | Euclidean motion (alignment-free) | Riemannian operations on groups |
The resulting principal components represent the major axes of shape variation within the sample, ordered by the amount of variance they explain. Each principal component corresponds to a mode of shape variation that can be visualized as a deformation from the mean shape. The PDM enables compact representation of shape variability through a limited number of principal components, facilitating statistical hypothesis testing, classification, and regression analysis of shape data.
Differential Coordinates Model
The Differential Coordinates model represents a more recent approach that addresses limitations of previous methods by employing a differential representation focused on local geometric variability [11]. This framework encodes shapes using differential coordinates that capture the local geometric structure of the shape, endowing the shape space with a Lie group structure that provides excellent theoretical properties and enables efficient algorithms [11]. Unlike the Point Distribution Model, this approach preserves the nonlinear nature of shape variation while offering computational advantages.
Mathematical Framework
In the Differential Coordinates model, shapes are represented using localized shape descriptors that are translation-invariant by construction [11]. The mathematical foundation leverages the fact that these differential coordinates form a Lie group, which provides:
- Closed-form expressions for Riemannian operations
- Numerical robustness without iterative approximation schemes
- Computational efficiency for geodesic calculations and interpolation
The model achieves rotational invariance through Procrustes alignment to a reference shape, similar to the Point Distribution Model, but preserves more of the nonlinear structure of shape variability [11]. This makes it particularly suitable for analyzing biological shapes with complex, nonlinear variation patterns that might be oversimplified by Euclidean approximation.
Experimental Protocol and Implementation
The implementation of Differential Coordinates analysis follows a structured workflow:
- Surface Mesh Representation: Shapes are represented as triangular surface meshes with vertices (v) and faces (f), where v ∈ ℝⁿˣ³ holds coordinates of n vertices and f ∈ ℝᵐˣ³ lists vertex indices forming m triangles [11]
- Reference Shape Selection: A representative template shape is chosen as the deformable reference
- Correspondence Establishment: Correspondences between vertices across all specimens are established (solving the "correspondence problem")
- Differential Coordinate Computation: Local shape descriptors are computed for each specimen
- Statistical Analysis: Geometric statistics are performed in the Lie group structure
Comparative Analysis and Applications
Geometric and Statistical Properties
Each shape framework offers distinct advantages for morphometric analysis. Kendall's shape space provides the most mathematically rigorous foundation with proper account of curvature but requires specialized geometric statistics [10]. The Point Distribution Model offers practical simplicity through linearization but may distort relationships in data with substantial shape variation [11]. The Differential Coordinates model balances computational efficiency with respect for nonlinear structure but requires more sophisticated implementation [11].
The curvature of shape spaces has important implications for statistical analysis. In Kendall's shape space, the intrinsic curvature means that linear combinations of shapes do not generally remain in the space, and averaging must be performed using Fréchet means [10]. The validity of tangent space approximation depends on the scale of variation in the dataset, with empirical evidence suggesting it works well for most biological applications where variation is relatively limited [10].
Applications in Morphometrics and Drug Development
These mathematical frameworks enable sophisticated analysis of biological shapes with applications in evolutionary biology, systematics, and increasingly in biomedical research and drug development. Specific applications include:
- Quantifying morphological changes in response to pharmaceutical treatments
- Characterizing disease progression through structural changes in tissues or organs
- Classifying pathological states based on cellular or subcellular morphology
- Analyzing anatomical development and the effects of genetic or environmental factors
In drug development, shape analysis can reveal subtle treatment effects that might be missed by traditional measurements, providing biomarkers for efficacy or toxicity. The probabilistic extensions of these frameworks, such as the Kendall Shape Probabilistic U-Net that incorporates shape spaces into deep learning models, further expand applications to image segmentation and analysis in biomedical imaging [12].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools for Shape Analysis
| Tool/Reagent | Function | Application Context |
|---|---|---|
| morphomatics Library | Implementation of shape space models | General shape analysis across frameworks |
| Surface Mesh (v, f) | Digital representation of shapes | Discrete representation of biological forms |
| Procrustes Alignment Algorithm | Remove non-shape variation | Preprocessing for all shape frameworks |
| Principal Components Analysis | Dimensionality reduction | Point Distribution Model implementation |
| Exponential/Logarithmic Maps | Geodesic calculations | Navigation in nonlinear shape spaces |
| Kendall Shape VAE | Probabilistic shape modeling | Shape-aware image segmentation [12] |
| PyVista | 3D visualization | Visualizing shapes and deformations [11] |
Kendall's Shape Space, Point Distribution Models, and Differential Coordinates provide complementary mathematical frameworks for analyzing biological shapes in morphometrics research. Kendall's approach offers a rigorous foundation based on Riemannian geometry, the Point Distribution Model enables practical application of multivariate statistics through linearization, and Differential Coordinates balance computational efficiency with respect for nonlinear structure. Together, these frameworks empower researchers to quantify and analyze morphological variation with unprecedented mathematical precision, opening new avenues for understanding biological form in contexts ranging from evolutionary biology to pharmaceutical development. As shape analysis continues to evolve, these core mathematical frameworks provide the foundation for increasingly sophisticated analysis of biological morphology and its relationship to function, development, and evolution.
The precise quantification of biological form is fundamental to understanding patterns of growth, evolution, and variation. Geometric morphometrics (GM) has emerged as the gold standard for analyzing shape, using coordinate-based data to quantify morphological differences while preserving geometric information throughout statistical analyses [13] [6]. This approach has transformed the study of phenotypic evolution by enabling researchers to capture and analyze complex anatomical structures with unprecedented precision. Shape representation in GM relies primarily on three complementary data types: landmarks, semilandmarks, and surface meshes, each addressing specific challenges in capturing biological form.
The fundamental challenge in morphometrics lies in establishing biological homology—ensuring that compared points represent the same biological entity across specimens. While landmarks provide discrete points of known homology, many biological structures lack sufficient such points for comprehensive shape characterization. This limitation has driven the development of semilandmarks and surface representations that densely sample curves and surfaces between traditional landmarks [14] [13]. These approaches have expanded the scope of morphometric studies to encompass entire structures rather than being limited to discrete points, enabling more nuanced investigations of morphological variation and evolution.
Landmarks: The Foundation of Geometric Morphometrics
Definition and Classification
Landmarks are defined as discrete, anatomically corresponding points that can be reliably identified across all specimens in a study. These points represent biological homologues, meaning they share common evolutionary and developmental origins [14] [15]. In geometric morphometrics, landmarks are typically categorized into three distinct types based on their anatomical definability:
- Type I landmarks are defined by local biological features, such as sutures between bones or small foramina, where distinct structures intersect.
- Type II landmarks represent points of maximum curvature or other local geometric features that can be reliably located based on tissue morphology.
- Type III landmarks are defined extrinsically, often as extremal points that require reference to other landmarks or the specimen's boundaries.
The primary strength of landmarks lies in their established biological homology, which provides a solid foundation for interpreting statistical shape differences in evolutionary or developmental contexts [14]. This biological validity makes them indispensable for studies investigating transformational processes.
Limitations and Constraints
Despite their biological relevance, landmarks present significant practical limitations. Their number is constrained by the availability of clearly identifiable homologous points, which rapidly diminishes when studying closely related taxa or smooth biological surfaces [13]. This problem becomes particularly acute in phylogenetic broad-scale studies, where identifiable homologous points become increasingly scarce [6]. Furthermore, landmarks alone cannot capture the morphological information between discrete points, potentially missing substantial shape variation occurring across curves and surfaces [13].
Table 1: Landmark Types and Their Characteristics in Morphometric Analysis
| Landmark Type | Definition Basis | Biological Homology | Examples | Primary Limitations |
|---|---|---|---|---|
| Type I | Local biological features | Strong | Sutures, foramina | Limited number on smooth surfaces |
| Type II | Maximum curvature | Moderate | Bony processes, apex of curves | More susceptible to identification error |
| Type III | Extrema relative to other points | Weak | Extremal points, endpoints | Most dependent on overall configuration |
Semilandmarks: Enhancing Shape Capture
Conceptual Framework
Semilandmarks (also called "sliding semilandmarks") were developed to address the limitation of sparse landmark coverage by providing a method to quantify shape along curves and surfaces between traditional landmarks [13]. Unlike landmarks, semilandmarks do not possess established biological homology in the traditional sense. Instead, they rely on geometric homology, where equivalence is determined algorithmically based on their relative positions on curves or surfaces bounded by true landmarks [14] [15].
The theoretical foundation of semilandmarks recognizes that while individual semilandmark positions may not be biologically meaningful, the overall curves and surfaces they represent are homologous structures [15]. As noted by researchers, "the coordinates of semilandmarks along the surface are meaningless, and one cannot interpret the position of single semilandmarks, only the surface geometry that all semilandmarks describe together" [15]. This conceptual shift requires treating semilandmarks as a collective representation of form rather than as discrete homologous points.
Technical Implementation
The placement of semilandmarks follows a multi-stage process. First, a template specimen is manually landmarked, and semilandmarks are distributed along curves or across surfaces between landmarks. This template is then warped to each target specimen using thin-plate spline (TPS) interpolation based on the true landmarks [14] [16]. The semilandmarks are subsequently "slid" to minimize either bending energy or Procrustes distance, effectively removing the tangential component of their placement error [14] [13].
Two primary criteria are used for the sliding process:
- Bending energy minimization: This approach positions semilandmarks to minimize the deformation energy required to transform the template to the target specimen, giving greater weight to landmarks and semilandmarks local to the point being slid [14].
- Procrustes distance minimization: This method slides semilandmarks to minimize the squared Procrustes distance between specimens, where all landmarks and semilandmarks influence the sliding equally, regardless of proximity [14].
The number of iterations in the sliding process affects the final configuration, with research indicating that classification accuracy stabilizes after approximately 12 iterations rather than progressively improving with more iterations [16].
Surface Meshes and Advanced Approaches
Surface Mesh Representation
Surface meshes provide the most comprehensive approach to shape representation by capturing continuous anatomical surfaces rather than discrete points. A surface mesh consists of vertices (points), edges (connections between points), and faces (polygonal surfaces, typically triangles), creating a continuous representation of the anatomical structure [15]. Surface meshes are particularly valuable for visualizing statistical results, as they can be warped to landmark and semilandmark configurations to create realistic representations of mean shapes or shape extremes [15].
In practical application, a template surface mesh is warped to fit estimated landmark and semilandmark configurations using thin-plate spline interpolation [15]. This enables the creation of surfaces representing statistical estimates, such as means or allometrically scaled shapes, which have utility in clinical contexts for assessing anomalies or building models for functional analyses like finite element analysis [15].
Landmark-Free Methods
Recent technological advances have prompted the development of landmark-free approaches that bypass traditional landmark identification entirely. These methods include:
- Deterministic Atlas Analysis (DAA): A Large Deformation Diffeomorphic Metric Mapping (LDDMM) approach that computes deformations between a dynamically generated atlas and each specimen without requiring pre-defined landmarks [6].
- Iterative Closest Point (ICP) algorithms: Rigid registration approaches that align surfaces by iteratively minimizing distances between point clouds [14] [6].
- Auto3dgm: A package that uses a template specimen with the greatest geometric similarity to project semilandmarks to all other specimens [14].
These landmark-free methods offer significant advantages in processing speed and reduced researcher bias, making them particularly suitable for analyzing large datasets [6]. However, they face challenges when applied to phylogenetically disparate taxa, as the correspondence points identified may not represent biological homologues [6].
Table 2: Comparison of Semilandmarking and Landmark-Free Approaches
| Method | Basis of Correspondence | Homology Assurance | Automation Level | Best Application Context |
|---|---|---|---|---|
| Sliding Semilandmarks | Landmark-guided | Geometric homology | Semi-automated | Studies requiring biological interpretability |
| Deterministic Atlas Analysis | Deformation-based | Sample-dependent | Automated | Large-scale studies across disparate taxa |
| Iterative Closest Point | Surface proximity | Topographic similarity | Automated | Classification and discrimination tasks |
| Auto3dgm | Template projection | Geometric similarity | Automated | Rapid data processing |
Methodological Comparisons and Experimental Insights
Performance of Different Semilandmarking Approaches
Comparative studies have systematically evaluated the performance of different semilandmarking approaches, revealing both consistencies and important differences. One comprehensive study compared three landmark-driven approaches: sliding TPS, hybrid rigid registration combining least-squares and ICP algorithms (LS&ICP), and an approach combining TPS with non-rigid ICP (TPS&NICP) [14] [15]. The findings demonstrated that while sliding TPS and TPS&NICP produced highly consistent results, the LS&ICP approach yielded notably different semilandmark locations and subsequent statistical outcomes [15].
These differences translated to variations in estimates of mean shapes, principal components of shape variation, and allometric patterns [14] [15]. Importantly, consistency within methods was highest for sliding TPS and TPS&NICP, particularly when true landmarks were densely distributed across the surface [15]. This suggests that the performance of semilandmarking approaches is contingent on the landmark framework guiding them.
Effect of Semilandmark Density
The density of semilandmarks represents a critical methodological decision in study design. Research indicates that while increasing semilandmark density enhances shape capture, it does not necessarily improve analytical outcomes proportionally. Studies comparing different densities found that estimates of surface mesh shape remained generally consistent across densities, suggesting that beyond a certain threshold, additional semilandmarks provide diminishing returns [15].
However, surfaces warped using landmarks alone demonstrated notable differences compared to those incorporating semilandmarks, with the discrepancy dependent on landmark coverage and template selection [15]. This underscores the importance of semilandmarks for accurately representing surfaces between landmarks, particularly in regions with sparse landmark coverage.
Experimental Protocol: Iteration Effects in Sliding Semilandmarks
A systematic investigation of iteration effects in sliding semilandmarks provides guidance for optimizing this parameter [16]. The experimental protocol employed the following methodology:
- Sample: 80 3D facial scans (40 males, 40 females) from the Stirling/ESRC 3D Face Database
- Template construction: 16 anatomical landmarks placed manually on a template mesh
- Semilandmark generation: 484 semilandmarks automatically generated and uniformly distributed across the facial surface
- Sliding procedure: Semilandmarks slid along target meshes using TPS warping with bending energy minimization
- Iteration test: Five relaxation states tested (1, 6, 12, 24, and 30 iterations)
- Analysis: Principal Component Analysis for feature selection followed by Linear Discriminant Analysis for gender classification
The results demonstrated that classification accuracy peaked at 12 iterations (96.43%) rather than increasing progressively with more iterations [16]. This indicates an optimal threshold for the sliding process beyond which additional iterations do not improve results and may even reduce accuracy. The processing time increased linearly with iteration count, making higher iterations computationally expensive without analytical benefit [16].
Practical Applications and Classification Context
Nutritional Status Assessment in Children
Geometric morphometrics has demonstrated significant utility in practical classification problems, such as assessing nutritional status in children. The SAM Photo Diagnosis App Program developed a smartphone application to identify severe acute malnutrition in children aged 6-59 months from images of their left arms [17] [18]. This approach represents an innovative application of GM techniques in real-world screening contexts.
The methodology involved:
- Sample collection: Images from 410 Senegalese children with either optimal nutritional condition or severe acute malnutrition
- Landmark and semilandmark placement: Anatomical points digitized on arm images
- Classification model development: Linear discriminant analysis applied to shape variables
- Out-of-sample validation: Methodological framework for classifying new individuals not included in the training sample
A critical challenge addressed in this research was the classification of out-of-sample individuals, requiring specialized approaches to obtain registered coordinates in the training sample's shape space [17]. This application highlights the translation of morphometric shape representation from theoretical framework to practical tool with significant public health implications.
Taxonomic Discrimination in Fisheries
Geometric morphometrics has also proven valuable in taxonomic discrimination, as demonstrated in a study of three Indian shad species [19]. The research analyzed digital images from 120 specimens using GM approaches to investigate body shape variations. The analysis successfully differentiated the species with 100% accuracy using Canonical Variate Analysis and Discriminant Function Analysis, though the limited sample size for one species (Hilsa kelee, n=6) necessitated leave-one-out cross-validation to address potential overfitting [19].
This application illustrates how shape representation using landmarks and semilandmarks can provide discrimination beyond traditional morphometric approaches, offering insights into subtle morphological differences with taxonomic significance.
Table 3: Essential Software Tools for Shape Representation in Morphometrics
| Software Tool | Primary Function | Key Features | Application Context |
|---|---|---|---|
| EVAN Toolbox | Semilandmark processing | Sliding semilandmarks along curves and surfaces | General morphometric analysis |
| Viewbox | Template creation and warping | Semiautomated semilandmark placement | 3D facial analysis [16] |
| Geomorph R Package | Statistical shape analysis | Procrustes ANOVA, phylogenetic integration | Comprehensive GM analysis |
| Morpho R Package | Sliding semilandmarks | Minimization of bending energy/Procrustes distance | Landmark and semilandmark processing |
| Deformetrica | Landmark-free analysis | Deterministic Atlas Analysis (DAA) | Large-scale datasets [6] |
| Auto3dgm | Automated correspondence | Template-based point correspondence | Rapid data processing [14] |
The representation of biological form through landmarks, semilandmarks, and surface meshes provides a sophisticated framework for analyzing shape variation within a defined shape space. Each approach offers distinct advantages: landmarks provide biological homology, semilandmarks enable dense shape capture between landmarks, and surface meshes facilitate comprehensive visualization and analysis. The integration of these methods allows researchers to construct detailed representations of morphological variation that can be leveraged for classification purposes.
Each methodological choice in shape representation carries implications for subsequent classification analyses. Landmark-based approaches maintain biological interpretability but may lack comprehensive shape coverage. Semilandmark methods enhance shape capture but introduce algorithmic dependence in point placement. Landmark-free approaches offer automation and efficiency but may sacrifice biological correspondence. Understanding these trade-offs is essential for designing morphometric studies that yield biologically meaningful and statistically robust classification systems.
As morphometric research continues to evolve, the integration of traditional landmark-based approaches with emerging landmark-free methods holds promise for addressing the challenges of analyzing increasingly large and complex morphological datasets. This synthesis will expand the scope of morphometric studies and enhance our understanding of shape variation across evolutionary, developmental, and clinical contexts.
The quantification of shape is a fundamental challenge across numerous scientific disciplines, from evolutionary biology and archaeology to modern drug discovery. Geometric morphometrics (GM) provides a powerful suite of tools for addressing this challenge by capturing and analyzing the geometry of anatomical structures or objects while controlling for differences in size, position, and orientation [20]. The core concept in GM is shape space—a mathematical space in which each point represents a unique object shape. Navigating this space requires robust metrics to quantify similarity and difference, enabling researchers to classify specimens, identify patterns of variation, and test hypotheses about form and function [17].
This technical guide focuses on two primary classes of tools for this task: Procrustes distance, which measures the difference between shapes after optimal superimposition, and multidimensional shape similarity metrics, which integrate numerous geometric descriptors to predict perceptual similarity. The Procrustes paradigm is particularly central to modern morphometrics, as it provides a rigorous framework for placing specimens into a common coordinate system for statistical analysis [20]. Understanding these metrics is essential for anyone working in morphometrics research, as they form the basis for almost all subsequent statistical analyses and interpretations of shape data.
Theoretical Foundations of Shape Similarity
The Challenge of Quantifying Shape
Quantifying visual shape similarity is a complex problem because shape perception involves multiple competing constraints. An effective shape representation must balance sensitivity (the ability to discriminate between subtly different shapes) and robustness (providing a consistent description across irrelevant transformations like rotation or scaling) [21]. Different shape descriptors inherently represent trade-offs between these goals; a descriptor invariant to rotation may be highly sensitive to other transformations like "bloating" or the addition of noise to a contour [21].
Human visual perception likely resolves this conflict by representing shape in a multidimensional space defined by many complementary shape descriptors [21]. This approach motivates computational models that combine multiple geometric properties to predict human shape similarity judgments. No single metric can perfectly capture all aspects of shape similarity, which is why the field employs a variety of distance measures tailored to different data types and research questions.
Procrustes Distance and Shape Space
The Procrustes distance is a cornerstone metric in geometric morphometrics for comparing shapes defined by landmark coordinates. The process begins with a set of homologous landmarks—anatomically corresponding points—captured from each specimen. The core idea of Procrustes analysis is to remove the effects of non-shape-related variation through an iterative least-squares optimization process known as Generalized Procrustes Analysis (GPA) [20] [17].
The GPA procedure involves three sequential steps:
- Translation: Each shape configuration is centered to a common origin (typically the centroid of the landmarks) to eliminate positional differences.
- Scaling: Configurations are scaled to a standard size (unit centroid size) to remove the effect of size.
- Rotation: Configurations are rotated to minimize the sum of squared distances between corresponding landmarks [20].
After this superimposition, the Procrustes distance between two shapes is calculated as the square root of the sum of squared differences between the coordinates of their corresponding landmarks [22]. The resulting aligned coordinates reside in a non-Euclidean space known as Kendall's shape space. For statistical analysis, shapes are typically projected into a linear tangent space where standard multivariate methods can be applied with acceptable accuracy [20].
Table 1: Key Distance Metrics in Geometric Morphometrics
| Metric Name | Data Type | Calculation | Key Properties | Primary Applications |
|---|---|---|---|---|
| Procrustes Distance | Landmark coordinates | Square root of summed squared coordinate differences after GPA | Invariant to position, scale, rotation; defines geometric shape space | Hypothesis testing of shape difference, morphological systematics [20] [22] |
| Mahalanobis Distance | Multivariate data (e.g., Procrustes coordinates) | Measures distance in terms of standard deviations from a group mean, accounting for covariance | Scale-invariant, accounts for variable correlations | Classifying specimens into groups, discriminant analysis [22] |
| ShapeComp Similarity | 2D contours/outlines | Multidimensional Euclidean distance from >100 shape features (e.g., area, compactness) | Predicts human perceptual similarity; perceptually uniform stimuli | Psychophysical research, visual neuroscience, AI vision [21] |
Methodological Protocols for Shape Analysis
A Standard Procrustes Analysis Workflow
The following workflow details the essential steps for a landmark-based geometric morphometric study, from data collection to statistical analysis. This protocol is adapted from applications in osteology [20] and entomology [22].
Step 1: Data Acquisition and Digitization
- Specimen Selection: Ensure specimens are well-preserved and represent the biological variation of interest. For bilateral structures, decide whether to use one side (flipping scans if necessary) or both.
- Landmark Definition: Establish a template of homologous landmarks (fixed anatomical points), curve semi-landmarks (points along homologous curves), and surface semi-landmarks (points on homologous surfaces). The number and placement of points are critical to capture morphology without over-sampling [20].
- Data Capture: Use high-resolution 3D scanners (e.g., structured-light scanners like Artec Eva) or 2D imaging systems. For 2D, ensure consistent orientation and scale. Save data as coordinate matrices.
Step 2: Configuration Preprocessing
- Data Organization: For
nspecimens, each withklandmarks inmdimensions (2D or 3D), combine coordinate matrices into ak x m x narray [20]. - Handling Missing Data: For damaged specimens, employ imputation methods. The choice of method (e.g., regression-based, thin-plate spline) depends on the extent of missingness [20].
- Semi-landmark Sliding: If used, relax semi-landmarks along curves and surfaces to minimize bending energy, optimizing their positions as homologous points.
Step 3: Generalized Procrustes Analysis (GPA)
- Translation: Center each configuration by subtracting its centroid (mean x, y, z coordinates).
- Scaling: Scale all configurations to unit Centroid Size (CS), calculated as the square root of the sum of squared distances of all landmarks from their centroid.
- Rotation: Rotate configurations to minimize the global sum of squared distances between corresponding landmarks via an iterative algorithm. The final output is the set of Procrustes-aligned coordinates.
Step 4: Statistical Analysis and Distance Calculation
- Procrustes ANOVA: Test for significant shape difference between groups, accounting for measurement error and other factors.
- Principal Component Analysis (PCA): Reduce dimensionality of aligned coordinates to visualize major trends of shape variation in a morphospace [22].
- Distance Calculation: Compute Procrustes distances between specimens or group means for clustering and classification. Use Mahalanobis distances for group discrimination [22].
Protocol for Out-of-Sample Classification
A common challenge in applied morphometrics is classifying a new specimen that was not part of the original Procrustes alignment. The following protocol addresses this [17]:
- Template Selection: Choose a single specimen or the mean shape from the reference (training) sample to serve as a template.
- Target Registration: Perform a Procrustes fit of the new specimen's raw coordinates to the selected template. This is a partial Procrustes alignment that removes non-shape variation relative to the template, without a full GPA involving all specimens.
- Projection: Project the newly registered coordinates into the existing tangent space of the reference sample.
- Classification: Apply the pre-existing classification rule (e.g., linear discriminant function) to the new specimen's projected coordinates.
Table 2: The Scientist's Toolkit: Essential Reagents and Software for Geometric Morphometrics
| Tool/Reagent | Specification/Type | Primary Function in Workflow |
|---|---|---|
| High-Resolution 3D Scanner (e.g., Artec Eva) | Hardware | Captures surface topography of specimens to create 3D digital models for landmarking [20]. |
| Digitization Software (e.g., Viewbox 4, TPSDig2) | Software | Provides interface for placing and recording coordinates of landmarks, curve points, and surface points on digital specimens [20] [22]. |
| Geometric Morphometrics Software (e.g., MorphoJ, R package geomorph) | Software | Performs core analyses: Generalized Procrustes Analysis, PCA, statistical testing of shape difference, and visualization [22]. |
| Statistical Environment (e.g., R) | Software | Provides a flexible platform for advanced statistical analysis, custom scripting, and data visualization of shape data [20] [17]. |
| Human Os Coxae Template [20] | Research Protocol | Pre-defined set of landmarks for a specific structure; ensures consistency and homology across studies. |
| Shape Feature Model (e.g., ShapeComp) [21] | Computational Model | Predicts human perceptual shape similarity from outlines using a multidimensional feature space; useful for psychophysics and AI. |
Applications and Case Studies in Research
Taxonomic Classification of Thrips
Geometric morphometrics successfully distinguishes closely related insect species where traditional methods struggle. A 2025 study on eight species of Thrips used 11 landmarks on the head and 10 on the thorax. Procrustes-based PCA revealed significant shape differences, with the first three principal components accounting for over 73% of head shape variation. Procrustes distance and Mahalanobis distance matrices, analyzed with permutation tests, statistically confirmed species separations. For instance, T. angusticeps and T. australis showed the greatest head shape divergence, while the thorax landmark configuration best separated T. nigropilosus, T. obscuratus, and T. hawaiiensis. This demonstrates GM's power as a complementary tool for identifying quarantine-significant pests [22].
Nutritional Status Assessment in Children
GM enables non-invasive nutritional screening by analyzing body shape. The SAM Photo Diagnosis App Program uses a smartphone app to classify nutritional status in children aged 6-59 months from photos of the left arm. A discriminant model is built from Procrustes-aligned landmarks and semi-landmarks from a reference sample. For out-of-sample classification, the app registers a new child's arm photo to a template from the reference sample, projecting it into the established shape space for classification. This digital health tool highlights GM's potential for real-world public health interventions, relying on a robust registration and classification protocol for new individuals [17].
Analyzing Morphological Integration in Human Osteology
A 2025 study of the human os coxae (hip bone) illustrates the use of Procrustes methods to investigate developmental and functional modularity. Researchers developed a detailed landmark template (25 fixed landmarks, 159 curve semi-landmarks, 425 surface semi-landmarks) from 3D scans. After Procrustes alignment, they analyzed patterns of shape covariation between the ilium, ischium, and pubis—bones that fuse during development. This protocol allowed them to test the hypothesis that these modules retain statistically independent patterns of variation due to their distinct developmental origins and functional roles, such as locomotion versus obstetric demands [20].
The fields of shape similarity quantification and geometric morphometrics are being transformed by the integration of artificial intelligence (AI) and machine learning (ML). In drug discovery, AI tools analyze the "chemical shape space" to perform virtual screening of millions of compounds, predicting bioactive molecules and optimizing lead compounds by assessing properties like shape similarity [23]. ML algorithms, including deep neural networks, are also being applied directly to morphometric data for classification tasks, potentially uncovering complex, non-linear patterns of shape variation that traditional methods might miss [17] [23].
Furthermore, advanced models like ShapeComp demonstrate that combining over 100 complementary shape descriptors (e.g., area, compactness, Fourier descriptors) into a single multidimensional metric can accurately predict human perceptual shape similarity, outperforming both pixel-based methods and some deep learning models [21]. This aligns with the core morphometric principle that no single metric can capture all aspects of shape, pointing toward a future of hybrid, multi-method approaches.
In conclusion, Procrustes distance provides a mathematically rigorous foundation for comparing shapes in a normalized space, while multidimensional similarity metrics offer powerful tools for modeling perceptual shape space. Together, these methodologies for quantifying shape similarity form an indispensable toolkit for modern morphometrics research. They enable the rigorous testing of hypotheses across diverse fields, from taxonomy and paleontology to biomedical engineering and drug discovery, driving forward our understanding of the relationship between form and function.
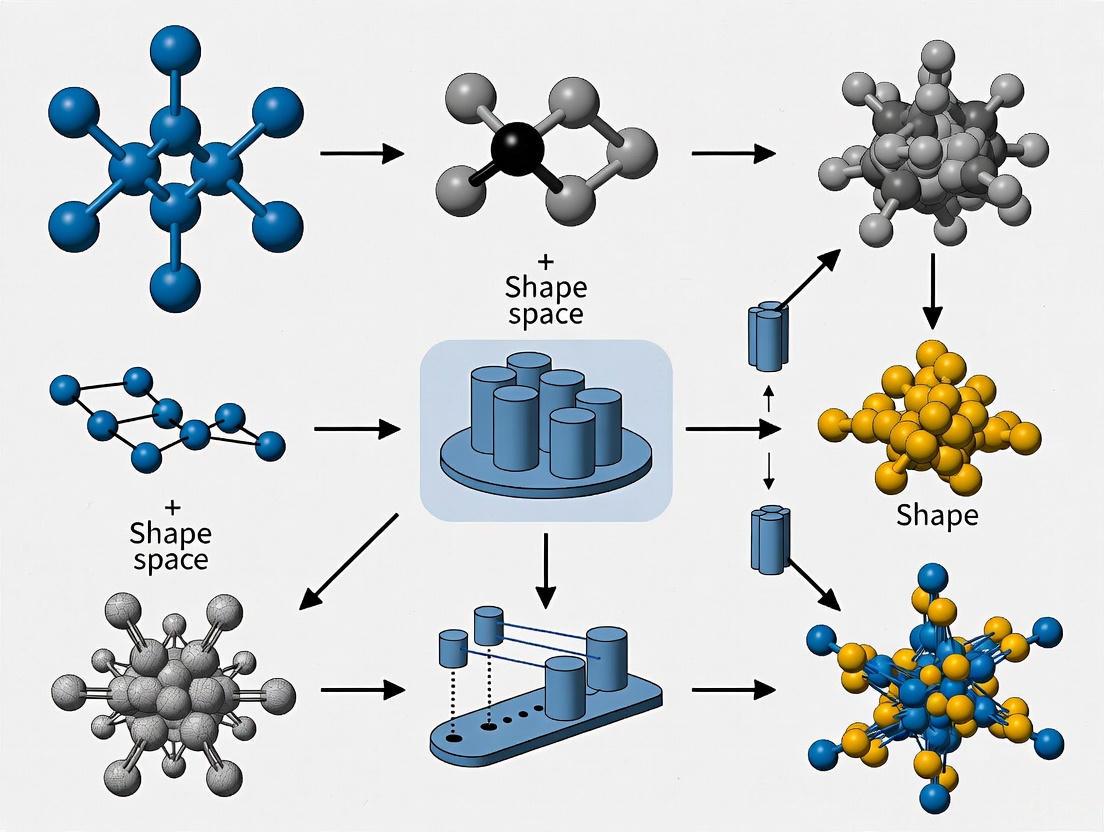
The quantification of biological shape is a cornerstone of evolutionary biology, medical imaging, and comparative anatomy. At the heart of this quantification lies the mapping problem—the challenge of establishing accurate, biologically meaningful correspondences between points on two or more anatomical structures. Whether comparing mammalian skulls across evolutionary timescales, analyzing differences in leaf morphology, or tracking morphological changes in medical conditions, researchers must solve this fundamental problem before any meaningful statistical analysis of shape can proceed [6]. The correspondence solution directly determines which aspects of shape variation are captured and ultimately influences all subsequent biological interpretations.
Traditional geometric morphometrics has largely relied on manual landmark placement—expert-identified homologous points that correspond across specimens. While this approach has proven immensely valuable, it introduces significant limitations: the process is time-consuming, susceptible to observer bias, and fundamentally constrained by the number of landmarks a researcher can practically place [24] [6]. As biological datasets expand to include thousands of 3D specimens obtained from CT scanning and other imaging technologies, and as research questions require more comprehensive capture of morphological detail, the field has increasingly turned toward automated correspondence methods that can operate without exhaustive manual intervention [24] [6]. These new approaches aim to capture shape variation more comprehensively while minimizing human bias, thereby enabling more powerful analyses of shape space and classification across diverse biological contexts.
Mathematical Foundations of Shape Correspondence
The mathematical treatment of shape correspondence has evolved along several parallel tracks, each with distinct advantages for particular biological applications. Quasi-conformal theory provides a powerful framework for representing surface deformations through Beltrami coefficients (μ), which quantify the local deviation from angle preservation. Intuitively, while conformal maps transform infinitesimal circles into infinitesimal circles, quasi-conformal maps transform them into ellipses with bounded eccentricity, providing a continuous measure of local distortion [25]. This formalism enables the computation of landmark-matching mappings between surfaces even when they lack global one-to-one correspondence, automatically detecting and aligning only the most relevant corresponding parts between two anatomical structures [25].
Diffeomorphic mapping approaches, particularly Large Deformation Diffeomorphic Metric Mapping (LDDMM), model shape transformations as smooth, invertible deformations that preserve topological structure. In methods like Deterministic Atlas Analysis (DAA), a mean template shape (an "atlas") is computed from the dataset, and the deformation required to map this atlas onto each specimen is quantified through momentum vectors ("momenta") at control points [6]. These momenta capture the optimal deformation trajectory and serve as the basis for comparing shape variation across specimens without requiring predefined landmarks.
Functional maps represent a more recent approach that operates in the spectral domain rather than directly in coordinate space. This method establishes correspondence through linear operators that map functions defined on one surface to another, effectively transforming the correspondence problem into one of finding a consistent basis between shapes [24]. The morphVQ pipeline leverages this approach with learned shape descriptors to estimate functional correspondence between whole triangular meshes, producing Latent Shape Space Differences (LSSDs) that characterize morphological variation through area-based and conformal operators [24].
Comparative Analysis of Correspondence Methodologies
Table 1: Key Methodologies for Solving the Shape Correspondence Problem
| Method | Mathematical Foundation | Correspondence Type | Key Advantages | Limitations |
|---|---|---|---|---|
| Traditional Landmarking | Procrustes superimposition | Discrete point homology | Biologically interpretable; well-established statistics | Limited morphological coverage; observer bias; time-intensive |
| morphVQ [24] | Functional maps + descriptor learning | Continuous surface mapping | Automated; captures comprehensive shape variation; computationally efficient | Requires quality surface meshes; black-box nature of learned descriptors |
| DAA (LDDMM) [6] | Diffeomorphic transformations | Deformation-based momentum vectors | No predefined landmarks needed; handles substantial shape differences | Sample-dependent atlas; sensitive to kernel width parameter; mixed modalities problematic |
| Quasi-conformal Registration [25] | Beltrami equation + quasi-conformal theory | Landmark-guided surface mapping | Handles inconsistent regions; optimal part-matching without global correspondence | Requires some landmark constraints; complex implementation |
| Auto3DGM [24] | Farthest point sampling + GDPF | Pseudolandmark correspondence | Fully automated; no template required | Lower resolution than surface-based methods |
Table 2: Performance Comparison on Biological Classification Tasks
| Method | Classification Accuracy | Computational Efficiency | Morphological Coverage | Required Expertise |
|---|---|---|---|---|
| Manual Landmarking | High (with expert digitization) | Low (hours to days per specimen) | Limited to landmark regions | High (domain knowledge required) |
| morphVQ [24] | Comparable to manual landmarking | High | Comprehensive (whole surfaces) | Medium (parameter tuning) |
| DAA [6] | Varies across taxa | Medium | Comprehensive | Medium (atlas selection critical) |
| Global PCA Models [26] | Moderate for gross morphology | High | Global geometry only | Low |
| Local/Wavelet Models [26] | High for detailed structures | Medium | Multi-scale detail | Medium |
The performance comparison reveals significant trade-offs between methodological approaches. morphVQ demonstrates particular strength in computational efficiency while maintaining classification accuracy comparable to manual landmarking [24]. DAA shows excellent potential for broad taxonomic comparisons but exhibits sensitivity to data preparation, particularly in handling mixed imaging modalities (CT vs. surface scans), though this can be mitigated through Poisson surface reconstruction to create watertight meshes [6]. Quasi-conformal registration excels in datasets where specimens share only partial correspondence, automatically identifying and aligning only common regions while excluding inconsistent parts [25].
Experimental Protocols for Correspondence Establishment
morphVQ Pipeline for Automated Phenotyping
The morphVQ pipeline implements a fully automated approach to shape correspondence through several refined stages. The process begins with data preparation and preprocessing, requiring triangular mesh models of biological specimens derived from micro-CT or other scanning modalities [24].
Step 1: Initial rigid alignment
- Apply the Generalized Dataset Procrustes Framework (GDPF) from auto3DGM
- Subsample shapes at low resolution (128-256 pseudolandmarks)
- Establish initial rotation and translation parameters for coarse alignment [24]
Step 2: Descriptor learning and functional map computation
- Learn shape descriptors directly from the aligned polygon models
- Estimate functional correspondences between pairs of specimens
- Use Consistent ZoomOut refinement to improve map quality [24]
Step 3: Latent Shape Space Difference (LSSD) calculation
- Compute area-based and conformal (angular) LSSDs
- These represent shape variation between specimens in the functional domain
- Enable statistical analysis of morphological variation [24]
Validation: The method has been validated through genus-level classification tasks, demonstrating comparable accuracy to manual landmarking while capturing more comprehensive morphological detail [24].
Deterministic Atlas Analysis (DAA) Protocol
DAA provides an alternative landmark-free approach suitable for datasets with substantial morphological variation. The protocol involves both preprocessing and analytical stages [6].
Preprocessing and standardization
- Convert all specimens to watertight, closed surfaces using Poisson surface reconstruction
- This critical step eliminates issues arising from mixed imaging modalities
- Ensure consistent mesh topology across the dataset [6]
Template selection and atlas generation
- Select an initial template specimen (choice has minimal impact on results)
- Iteratively compute a geodesic mean shape (atlas) that minimizes total deformation energy
- Generate control points guided by a kernel width parameter (e.g., 20.0mm produces ~270 control points) [6]
Deformation quantification
- Compute momentum vectors ("momenta") at each control point
- These represent optimal deformation trajectories from atlas to each specimen
- Perform kernel Principal Component Analysis (kPCA) on momenta to visualize shape space [6]
Parameter optimization: Kernel width selection balances morphological sensitivity with computational burden; smaller values (10.0mm) capture finer details but increase control points (1,782), while larger values (40.0mm) provide broader characterization with fewer points (45) [6].
DAA Experimental Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Tools for Shape Correspondence Research
| Tool/Software | Primary Function | Methodology | Application Context |
|---|---|---|---|
| morphVQ [24] | Automated shape correspondence | Functional maps + descriptor learning | General anatomical structures; bone morphology |
| Deformetrica [6] | Diffeomorphic registration | LDDMM/Deterministic Atlas Analysis | Macroevolutionary studies; disparate taxa |
| Geomorph [27] | Geometric morphometrics analysis | Traditional & modern GM | General biological shapes; comprehensive stats |
| MorphoLeaf [27] | Plant leaf morphometrics | Landmark & outline analysis | Plant leaves; digital identification |
| Auto3DGM [24] | Automated pseudolandmarking | Farthest point sampling + GDPF | General 3D shapes; initial alignment |
Visualization Framework for Correspondence Analysis
Shape Correspondence Method Classification
The solution to the mapping problem between shapes represents more than a technical exercise in computational geometry—it fundamentally shapes our understanding of biological form and its evolution. As correspondence methods evolve from discrete landmark-based approaches toward continuous, automated frameworks, they enable researchers to address more complex questions about morphological adaptation, diversification, and development across broader taxonomic scales. The emerging synergy between mathematical theory, computational implementation, and biological application promises to transform morphometrics from a specialized methodology into a general framework for understanding the evolution of form.
Each correspondence method carries implicit assumptions about the nature of biological variation, and the choice of method should be guided by the specific research question, dataset characteristics, and analytical goals. Landmark-based approaches retain value for hypothesis-driven studies of specific morphological structures, while landmark-free methods excel in exploratory analyses across disparate taxa or when comprehensive shape characterization is required. Future developments will likely focus on hybrid approaches that leverage the biological interpretability of landmarks with the comprehensive coverage of continuous correspondence methods, ultimately providing richer representations of shape space for classifying and understanding biological diversity.
From Theory to Practice: Classification Methods and Real-World Applications
In morphometrics research, the quantitative analysis of form and shape is fundamental to understanding biological variation, evolutionary patterns, and diagnostic characteristics in fields ranging from drug development to paleontology [28] [29]. The concept of "shape space" provides a mathematical framework where biological forms can be represented as points, enabling statistical analysis of morphological patterns that are often invisible to the human eye. Within this conceptual space, classification techniques serve as critical tools for identifying, categorizing, and interpreting complex morphological data. This technical guide provides an in-depth examination of three powerful classification methodologies—Linear Discriminant Analysis (LDA), Support Vector Machines (SVM), and Neural Networks—with specific emphasis on their application to morphometric research problems.
The drive toward more quantitative, reproducible, and objective analysis in morphology has accelerated the adoption of these machine learning techniques [29]. Traditional morphometric approaches often grapple with challenges of subjective interpretation and observer bias, limitations that can significantly impact research outcomes in pharmaceutical development and systematic biology. By contrast, LDA, SVM, and neural networks offer data-driven frameworks for morphological classification that can identify subtle, diagnostically significant patterns within high-dimensional shape data [30] [29]. This whitepaper examines the theoretical foundations, practical implementation, and relative strengths of these three techniques within the specific context of morphometric analysis.
Theoretical Foundations of Classification Techniques
Linear Discriminant Analysis (LDA)
Linear Discriminant Analysis is a supervised classification approach that operates by finding linear combinations of features that best separate two or more classes of objects or events [31] [32]. Developed from Fisher's linear discriminant in the 1930s, LDA follows a generative model framework, modeling the data distribution for each class and using Bayes' theorem to classify new data points [31]. The algorithm fundamentally seeks to identify a lower-dimensional projection that maximizes between-class variance while minimizing within-class variance, effectively enhancing class separability in the reduced space.
The core mathematical objective of LDA is to find the projection vector v that maximizes Fisher's criterion:
J(v) = (vᵀSᵇv) / (vᵀSʷv)
Where Sᵇ is the between-class scatter matrix and Sʷ is the within-class scatter matrix [31] [32]. For implementation, LDA operates under several key assumptions: the input data should follow a Gaussian distribution, the dataset should be linearly separable, and each class should share a common covariance matrix [31]. When these assumptions are met, LDA produces optimal classification boundaries with computational efficiency particularly valuable for high-dimensional morphological data where the number of features often exceeds sample size.
Support Vector Machines (SVM)
Support Vector Machines represent a distinct approach to classification, focusing on finding the optimal hyperplane that maximizes the margin between classes in a high-dimensional feature space [33] [34]. Developed in the 1990s, SVMs employ a discriminative approach, concentrating specifically on the instances most difficult to classify—the support vectors—which are the data points closest to the decision boundary [33] [35].
The fundamental optimization problem for a linear SVM can be expressed as:
minimize(½||w||² + C∑ζᵢ) subject to yᵢ(wᵀxᵢ + b) ≥ 1 - ζᵢ and ζᵢ ≥ 0
Where w is the normal vector to the hyperplane, C is a regularization parameter controlling the trade-off between maximizing margin and minimizing classification error, and ζᵢ are slack variables that allow for misclassification in non-separable cases [33]. For non-linearly separable data, SVMs employ the "kernel trick," mapping input features into higher-dimensional spaces using kernel functions such as Radial Basis Function (RBF), polynomial, or sigmoid kernels without explicitly computing the coordinates in that space [33] [34]. This capability makes SVMs particularly valuable for complex morphological patterns where linear separation is insufficient.
Neural Networks
Neural Networks, particularly deep learning architectures, represent a paradigm shift in classification capability through their ability to automatically learn hierarchical feature representations directly from raw data [28] [30]. Unlike LDA and SVM which typically operate on pre-engineered features, neural networks can discover and optimize the feature representation itself, making them exceptionally powerful for image-based morphometric analysis.
Convolutional Neural Networks (CNNs), the dominant architecture for image processing, employ a series of convolutional layers that progressively detect increasingly complex patterns—from edges and textures in early layers to sophisticated morphological structures in deeper layers [28] [30]. This hierarchical feature learning is achieved through multiple processing layers with learnable parameters, trained via backpropagation to minimize a loss function between predicted and actual classifications. For morphometric applications, this means CNNs can discern subtle shape characteristics that may be challenging to capture with traditional measurement-based approaches [28] [36].
Table 1: Core Mathematical Properties of Classification Techniques
| Technique | Optimization Objective | Decision Boundary | Key Parameters |
|---|---|---|---|
| LDA | Maximize between-class to within-class variance ratio | Linear | Number of components, prior probabilities |
| SVM | Maximize margin between classes | Linear or non-linear (via kernels) | Regularization C, kernel type, kernel parameters (e.g., γ for RBF) |
| Neural Networks | Minimize loss function via gradient descent | Highly non-linear | Network architecture, learning rate, number of epochs, batch size |
Comparative Technical Performance in Morphometric Research
Quantitative Performance Metrics
Recent research applications provide compelling evidence of the relative performance of these classification techniques in morphometric contexts. In archaeobotanical studies comparing domesticated and wild plant varieties, CNNs significantly outperformed traditional morphometric methods, achieving high classification accuracy even with limited training data [28]. Similarly, in taphonomic research analyzing carnivore tooth marks, CNNs achieved 81% classification accuracy compared to less than 40% for geometric morphometric methods including LDA-based approaches [36].
Table 2: Performance Comparison in Morphometric Applications
| Application Domain | LDA Performance | SVM Performance | Neural Network Performance | Reference |
|---|---|---|---|---|
| Archaeobotanical Identification | Not reported | Not reported | Beat outline analysis (EFT) in most cases, even with small datasets | [28] |
| Tooth Mark Classification | <40% accuracy | Not reported | 81% accuracy (DCNN), 79.52% (Few-Shot Learning) | [36] |
| Mesenchymal Stem Cell Analysis | Not primary method | Not primary method | 64% of studies used CNNs; up to 97.5% accuracy | [30] |
Morphometric Applications and Case Studies
The application of these classification techniques spans diverse morphometric research contexts. In archaeobotany, researchers have successfully employed CNNs to identify pairs of plant taxa using seed and fruit stone images, crucial for understanding domestication history [28]. Similarly, in paleontology, machine learning methods have demonstrated remarkable capability in fossil identification and taxonomic classification, overcoming long-standing challenges of observer bias and subjective interpretation [29].
Medical and pharmaceutical applications further illustrate the power of these techniques. In mesenchymal stem cell (MSCs) research, CNNs have become the dominant approach for tasks including cell classification (20% of studies), segmentation and counting (20%), and differentiation assessment (32%) [30]. These applications highlight how neural networks can automate image analysis while eliminating subjective biases, ultimately enhancing reproducibility in critical drug development contexts.
Implementation Protocols for Morphometric Classification
LDA Implementation Workflow
Implementing LDA for morphometric classification follows a structured protocol:
Data Preprocessing: Normalize and center the feature data, ensuring features are on comparable scales [31] [32]. For shape data, this may include Procrustes alignment for landmark-based morphometrics.
Feature Selection: Identify the morphometric descriptors (landmarks, outline coordinates, or other shape representations) that will serve as input features.
Model Training: Compute the between-class and within-class scatter matrices, then derive the linear discriminants by solving the generalized eigenvalue problem [31] [32].
Dimensionality Reduction: Project the original feature space onto the selected linear discriminants, typically reducing to k ≤ c-1 dimensions where c is the number of classes.
Classification: Apply Bayes' theorem in the reduced-dimensional space to assign class membership based on posterior probabilities [31].
SVM Implementation Protocol
The implementation of SVM for morphometric analysis requires careful consideration of data characteristics:
Data Preparation: Split morphometric data into training and testing sets, ensuring representative sampling across classes [34]. For shape data, consider feature standardization.
Kernel Selection: Choose an appropriate kernel function based on data separability:
Parameter Tuning: Employ grid search with cross-validation to optimize hyperparameters:
Model Training: Solve the quadratic optimization problem to identify support vectors and define the decision boundary [33].
Evaluation: Assess classification performance using metrics appropriate for morphometric research, potentially including precision, recall, and confusion matrix analysis [34].
Neural Network Implementation for Morphometric Analysis
Implementing neural networks for shape classification involves distinct considerations:
Data Preparation and Augmentation: For image-based morphometrics, apply transformations (rotation, scaling, translation) to increase dataset diversity and improve model robustness [28]. This is particularly valuable for small paleontological or archaeological datasets.
Architecture Selection: Choose an appropriate network architecture:
- Custom CNN for domain-specific morphometric problems
- Pre-trained networks (VGG19, ResNet) with transfer learning for limited data [28]
- Fully connected networks for landmark-based shape data
Training with Validation: Implement iterative training with separate validation monitoring to prevent overfitting, employing techniques like early stopping and dropout [28] [30].
Interpretation: Utilize activation maps and feature visualization to understand which morphological characteristics drive classification decisions, adding interpretability to predictions [30].
Research Reagent Solutions for Morphometric Classification
Implementing these classification techniques requires both computational and domain-specific tools. The following table outlines essential "research reagents" for morphometric classification studies:
Table 3: Essential Research Reagents for Morphometric Classification
| Reagent Category | Specific Tools/Solutions | Function in Morphometric Classification |
|---|---|---|
| Software Libraries | Scikit-learn, Momocs, Keras/TensorFlow, PyTorch | Provide implemented algorithms for LDA, SVM, and neural networks with optimized computational efficiency [31] [28] [32] |
| Data Acquisition Tools | Digital microscopes, CT scanners, outline digitization software | Capture high-fidelity morphological data for analysis [28] [36] |
| Shape Representation Methods | Elliptical Fourier Transforms (EFT), landmark coordinates, geometric morphometrics | Convert physical forms into quantitative data amenable to classification algorithms [28] [36] |
| Validation Frameworks | Cross-validation protocols, confusion matrix analysis, precision-recall metrics | Ensure methodological rigor and reproducible classification outcomes [31] [29] |
| Computational Infrastructure | GPU acceleration, cloud computing platforms | Handle computationally intensive training processes, particularly for deep learning applications [28] [30] |
Integrated Analysis Framework for Morphometric Classification
Technique Selection Guidelines
Choosing among LDA, SVM, and neural networks requires careful consideration of research constraints and objectives:
Select LDA when working with linearly separable morphometric data, when interpretability is paramount, when datasets are limited, or when computational resources are constrained [31] [32]. LDA performs optimally when its statistical assumptions are met.
Choose SVM for complex shape classification problems with clear margins between classes, for high-dimensional feature spaces, or when working with datasets where the number of features exceeds sample size [33] [35] [34]. SVM is particularly valuable when using non-linear kernels for complex morphological boundaries.
Employ Neural Networks for image-based morphometrics without clear feature representations, for very large and diverse datasets, or when maximum classification accuracy is the primary objective [28] [30] [36]. CNNs excel at discovering discriminative features directly from pixel data.
Emerging Trends and Future Directions
The field of morphometric classification is rapidly evolving, with several significant trends shaping research applications. Integrated approaches that combine traditional morphometric methods with machine learning are demonstrating particular promise [28] [36]. For instance, using outline analyses for feature extraction followed by neural networks for classification leverages the strengths of both approaches.
Methodological challenges remain, including the need for standardized validation frameworks and addressing the "black box" nature of complex models [30] [29]. Future developments will likely focus on explainable AI techniques to enhance interpretability, few-shot learning methods to address data scarcity common in morphometric research, and three-dimensional analysis frameworks that capture complete topographical shape information [36].
The application of LDA, SVM, and neural networks has fundamentally transformed morphometric research, enabling more quantitative, reproducible, and insightful analysis of biological form across diverse domains from pharmaceutical development to evolutionary biology. Each technique offers distinct advantages: LDA provides computational efficiency and interpretability, SVM delivers robust performance with complex decision boundaries, and neural networks offer unparalleled accuracy for image-based classification. As morphometric research continues to evolve toward more integrated, multi-method frameworks, understanding the theoretical foundations, implementation protocols, and relative strengths of these classification techniques becomes increasingly essential for researchers navigating the complex landscape of shape space analysis. The continued refinement of these methods promises to further enhance our ability to extract meaningful biological insights from morphological data, advancing both basic science and applied applications in drug development and beyond.
Alignment-Based vs. Alignment-Free Shape Similarity Methods
The quantitative analysis of shape, or morphometrics, is a cornerstone of modern biological research, enabling the precise characterization of form in fields ranging from evolutionary biology to drug discovery. At the heart of morphometrics lies the fundamental challenge of quantifying shape similarity—determining how to measure and compare the geometrical properties of biological structures while excluding non-shape variations such as size, position, and orientation. Two fundamentally different computational philosophies have emerged to address this challenge: alignment-based methods, which rely on establishing explicit point-to-point correspondences between shapes, and alignment-free methods, which compare shapes through abstract numerical descriptors without requiring explicit correspondence. Understanding the relative strengths, limitations, and applications of these approaches is essential for navigating shape space—the abstract mathematical space where each point represents a distinct shape configuration. This whitepaper provides a comprehensive technical comparison of these methodologies, framed within the context of shape classification and analysis in morphometric research.
Theoretical Foundations: Shape Spaces and Coordinate Systems
The concept of a shape space provides a rigorous mathematical foundation for morphometric analysis. A shape space is a multidimensional space in which each point corresponds to a unique shape configuration, and distances between points represent the magnitude of shape difference [10]. The structure of these spaces is complex and often non-Euclidean, creating both opportunities and challenges for shape analysis.
Kendall's Shape Space: This influential framework represents the shape of an object defined by landmarks as a point on a high-dimensional spherical surface. For 2D configurations with
klandmarks, the shape space has2k-4dimensions, while for 3D configurations, the dimensionality is3k-7[10]. These dimensions account for the removal of non-shape parameters: in 2D, one dimension each for size and rotation, and two for translation; in 3D, one for size, three for rotation, and three for translation.Procrustes Distance: The most widely used metric in alignment-based methods is Procrustes distance, which quantifies shape difference through a three-step process: (1) scaling configurations to unit centroid size, (2) translating configurations to a common position, and (3) rotating configurations to optimal alignment [10]. The full Procrustes distance further refines this by allowing additional scaling to minimize the residual sum of squared distances between corresponding landmarks.
Tangent Space Approximation: Because shape spaces are curved manifolds, statistical operations are often performed in a linear tangent space projecting from a reference shape (typically the mean shape). This approximation is generally valid for biological datasets where shape variation is relatively small compared to the curvature of the shape space [10].
Alignment-Based Methods: Principles and Protocols
Core Principles
Alignment-based methods, often termed geometric morphometrics, compare shapes by first establishing homologous correspondence—matching biologically equivalent points—between specimens. These methods explicitly separate shape from non-shape parameters through a process known as Generalized Procrustes Analysis (GPA). The core assumption is that meaningful shape comparison requires biological correspondence, which must be defined by an expert or through automated landmarking systems that preserve biological homology [6] [37].
Experimental Protocol: Generalized Procrustes Analysis
The standard protocol for alignment-based shape analysis involves the following steps:
Landmark Digitization: Anatomical structures are represented by landmarks—discrete points that can be precisely located and correspond biologically across specimens. These are typically categorized as:
- Type I: Discrete juxtapositions of tissues
- Type II: Maxima of curvature
- Type III: Extremal points
Procrustes Superimposition:
- Step 1: Centering - Translate all configurations so their centroids (centers of gravity) coincide at the origin [10].
- Step 2: Scaling - Scale all configurations to unit centroid size, defined as the square root of the sum of squared distances of each landmark from the centroid [10].
- Step 3: Rotation - Rotate configurations to minimize the sum of squared distances between corresponding landmarks relative to a reference configuration (typically the mean shape) [10].
Shape Variable Extraction: The resulting Procrustes coordinates represent shape variables, with the non-shape variation (position, size, orientation) removed. These coordinates reside in a curved shape space but are typically projected to a tangent space for multivariate statistical analysis.
Statistical Analysis: Conduct multivariate analyses (PCA, discriminant analysis, regression) on the shape variables to test biological hypotheses about shape variation, allometry, or group differences [9].
Limitations and Challenges
Despite their biological interpretability, alignment-based methods face several challenges:
- Landmark Definition: Identifying truly homologous points becomes increasingly difficult for disparate taxa or structures lacking clear anatomical landmarks [6].
- Operator Bias: Manual landmarking is time-consuming and susceptible to intra- and inter-observer bias, affecting reproducibility [6].
- Incomplete Shape Capture: Discrete landmarks may fail to capture information from curves and surfaces between landmarks, though this can be partially addressed with semi-landmarks [6].
- Template Dependency: Placing landmarks on new specimens requires either including them in a new global alignment or registering them to an existing template, which introduces methodological complexities [37].
Alignment-Free Methods: Principles and Protocols
Core Principles
Alignment-free methods circumvent the need for explicit point correspondence by representing shapes through numerical descriptors that capture global or local geometrical properties. These methods transform shape comparison into a problem of comparing numerical vectors in a feature space, making them particularly valuable for high-throughput analyses or when homologous landmarks are difficult to define [6] [7].
Key Approaches and Descriptors
Table 1: Major Classes of Alignment-Free Shape Descriptors
| Descriptor Class | Examples | Underlying Principle | Advantages | Limitations |
|---|---|---|---|---|
| Atomic Distance-Based | USR (Ultrafast Shape Recognition) [7] | Distribution of atomic distances from four reference points (centroid, etc.) | Extremely fast; no alignment needed; screens ~55M conformers/second | Cannot distinguish enantiomers; no chemical typing |
| Surface-Based | Spherical Harmonics, 3D Zernike Descriptors [7] | Mathematical decomposition of molecular surface | Rotationally invariant; compact representation | May oversimplify complex surfaces |
| Gaussian Overlay-Based | ROCS (Rapid Overlay of Chemical Structures) [2] | Volume overlap of Gaussian molecular models | Direct volume comparison; handles flexibility | Sensitive to initial orientation |
| Differential Coordinates | Fundamental Coordinates Model [11] | Metric distortion and curvature as elements of Lie groups | Invariant under Euclidean motion; valid shape instances guaranteed | Computationally complex |
| Deformation-Based | DAA (Deterministic Atlas Analysis) [6] | Deformation energy to map an atlas to each specimen | Captures continuous shape variation; automated | Parameter sensitive (kernel width) |
Experimental Protocol: Deterministic Atlas Analysis (DAA)
DAA is a landmark-free approach based on Large Deformation Diffeomorphic Metric Mapping (LDDMM) that has shown promise for macroevolutionary analyses [6]:
Atlas Generation:
- Select an initial template mesh from the dataset
- Iteratively compute a geodesic mean shape (atlas) by minimizing the total deformation energy required to map it onto all specimens [6]
Control Point Placement:
- Generate control points initially distributed evenly around the atlas
- Adjust point density according to shape variability in the dataset
- Kernel width parameter controls spatial extent: smaller values (e.g., 10.0 mm) yield finer-scale deformations with more control points [6]
Momentum Calculation:
- For each control point, compute a momentum vector ("momenta") representing the optimal deformation trajectory for aligning the atlas with each specimen
- These momenta operate within a Hamiltonian framework derived from the velocity field of ambient space [6]
Shape Comparison:
- Compare shapes using the momentum vectors as shape variables
- Apply kernel Principal Component Analysis (kPCA) to visualize and explore covariation in the shape data [6]
Protocol for Molecular Shape Similarity (USR)
Ultrafast Shape Recognition (USR) provides a rapid method for molecular shape comparison [7]:
Reference Point Calculation:
- Compute four reference points for each molecule: molecular centroid (ctd), closest atom to centroid (cst), farthest atom from centroid (fct), and farthest atom from fct (ftf)
Distance Distribution Calculation:
- For each reference point, calculate the distribution of distances to all other atoms in the molecule
- Characterize each distribution using its first three moments (mean, variance, and skewness), resulting in a 12-dimensional shape descriptor
Similarity Quantification:
- Compare molecules by computing the Euclidean distance or correlation between their 12-dimensional descriptor vectors
- No molecular alignment or superposition is required
Comparative Analysis: Performance and Applications
Quantitative Performance Comparison
Table 2: Method Comparison Across Applications
| Application Domain | Alignment-Based Performance | Alignment-Free Performance | Key Findings |
|---|---|---|---|
| Virus Taxonomy Classification [38] | High accuracy but computationally expensive (ClustalW, MUSCLE, MAFFT) | K-merNV and CgrDft perform similarly to alignment methods | Encoded methods provide faster results suitable for large datasets or time-sensitive variant detection |
| Macroevolutionary Analysis (Mammals) [6] | Manual landmarking captures detailed homologous variation | DAA shows strong correlation after mesh standardization (Poisson reconstruction) | Both methods produced comparable but varying estimates of phylogenetic signal, disparity, and evolutionary rates |
| Molecular Virtual Screening [7] [2] | Limited application without known structure | USR, ROCS successfully identify active compounds; enable scaffold hopping | Shape-based methods effective for lead discovery; often outperform 2D similarity |
| Nutritional Assessment (Arm Shape) [37] | Effective within sample; complex for new individuals | Not directly applicable | Challenge in classifying out-of-sample individuals without re-alignment |
| Distal Radius Symmetry [39] | Limited to predefined landmarks | Landmark-free morphometry enables full surface analysis | Revealed strong intraindividual symmetry supporting contralateral template use |
Computational Efficiency
A critical advantage of alignment-free methods is their significantly reduced computational burden. For virus taxonomy classification, alignment-based methods like ClustalW and MUSCLE require pairwise comparison of all sequences, which becomes computationally prohibitive for large datasets [38]. In contrast, encoded methods like K-merNV represent sequences as numerical vectors, enabling rapid similarity computation through simple distance metrics [38]. Similarly, in molecular shape comparison, USR can screen millions of compounds per second, while alignment-based methods require iterative optimization of molecular superposition [7].
Biological Interpretability
While alignment-free methods often excel in computational efficiency, alignment-based approaches typically provide superior biological interpretability. The Procrustes coordinates from geometric morphometrics directly correspond to anatomical locations, allowing researchers to visualize shape changes as actual deformations of biological structures [10] [9]. This facilitates the interpretation of results in terms of specific biological processes or functional adaptations.
Integrated Workflow and Decision Framework
Method Selection Guide
The choice between alignment-based and alignment-free methods depends on multiple factors:
Choose Alignment-Based Methods When:
- Homologous points can be reliably identified across all specimens
- Biological interpretation of specific anatomical changes is required
- Sample sizes are manageable (hundreds rather than thousands)
- Comparing morphologically similar specimens
Choose Alignment-Free Methods When:
- Analyzing highly disparate taxa with few homologous points
- Processing large datasets (thousands of specimens)
- Computational efficiency is prioritized
- Dealing with molecular structures or complex surfaces
Hybrid Approaches
Emerging methodologies seek to combine the strengths of both approaches. For instance, landmark-free methods like DAA can establish dense correspondence without manual landmarking, then export landmark-like points for traditional morphometric analysis [6]. Similarly, in molecular sciences, hybrid workflows might use alignment-free methods for rapid screening followed by alignment-based analysis for detailed study of top candidates [7].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Software and Analytical Tools
| Tool Name | Method Category | Primary Function | Application Context |
|---|---|---|---|
| MEGA11 [38] | Alignment-Based | Multiple sequence alignment (ClustalW, MUSCLE) | Virus taxonomy, evolutionary genetics |
| NGphylogeny [38] | Alignment-Based | Online phylogenetic analysis (MAFFT, ClustalOmega) | Accessible phylogenetic reconstruction |
| Deformetrica [6] | Alignment-Free | DAA implementation using LDDMM | Macroscopic shape analysis (e.g., mammilian crania) |
| Morphomatics [11] | Both | Shape space analysis (Kendall, FundamentalCoords) | General morphometric research |
| ROCS [2] | Alignment-Free | Rapid molecular shape similarity | Virtual screening, drug discovery |
| USR-VS [7] | Alignment-Free | Ultrafast molecular shape screening | High-throughput virtual screening |
| MITK [39] | Data Preprocessing | Medical image segmentation and mesh extraction | Biomedical shape analysis |
The dichotomy between alignment-based and alignment-free methods for shape similarity analysis represents a fundamental trade-off in morphometrics: biological interpretability versus computational efficiency and automation. Alignment-based methods, rooted in Procrustes geometry and explicit homology, provide a biologically meaningful framework for shape analysis but face challenges in scalability and landmark identification for disparate forms. Alignment-free methods, leveraging numerical descriptors and deformation energies, offer powerful alternatives for high-throughput analysis and complex morphological systems where homology is obscure.
The future of shape analysis lies not in the dominance of one approach over the other, but in the continued development of hybrid methodologies that leverage the strengths of both paradigms. As landmark-free techniques improve their biological interpretability and alignment-based methods enhance their automation, the morphometrics community moves closer to comprehensive frameworks for navigating shape spaces across biological scales—from molecular structures to organismal forms. This integration will ultimately expand the scope of morphometric studies, enabling the analysis of larger and more diverse datasets while preserving the biological insights that make shape analysis fundamentally meaningful.
Visual Workflows
Alignment-Based Shape Analysis Workflow
Alignment-Free Shape Analysis Workflow
The concept of shape space, fundamental to geometric morphometrics (GM) research, provides a powerful framework for understanding molecular complementarity in drug discovery. In GM, complex biological forms are captured using coordinate points, superimposed through Procrustes alignment to remove differences in location, size, and orientation, and projected into a multidimensional shape space where statistical analyses reveal patterns of variation and covariation [20]. This precise mathematical approach to form analysis has direct parallels in computational drug discovery, where the three-dimensional shape of a molecule often determines its biological activity and binding affinity to protein targets.
Virtual screening using 3D shape similarity has emerged as a cornerstone of modern drug discovery, enabling researchers to rapidly identify potential drug candidates from chemical libraries containing billions of compounds. Rather than relying solely on two-dimensional structural similarity, these methods recognize that molecules with similar three-dimensional shapes often share similar biological properties, even if their underlying chemical scaffolds differ substantially [40] [41]. This principle of "scaffold hopping" – identifying structurally different compounds that maintain similar biological activity – is particularly valuable for designing novel therapeutics with improved efficacy and safety profiles [41].
This technical guide examines the core methodologies, applications, and experimental protocols for leveraging 3D shape similarity in virtual screening and target prediction, framing these computational approaches within the broader morphometric research context of shape space analysis and classification.
Theoretical Foundations: From Biological Form to Molecular Shape
Geometric Morphometrics in Drug Discovery
The Procrustean analytical protocol, widely used in geometric morphometrics, involves three fundamental operations: removing positional differences by centering configurations on a common origin, eliminating size differences through rescaling, and removing rotational effects through alignment [20]. When applied to molecular structures, this approach allows researchers to compare molecular shapes independent of their orientation or overall dimensions, focusing instead on the spatial arrangement of key functional elements.
The mathematical foundation for this approach lies in Kendall's shape space, a non-Euclidean space where molecular configurations are represented after normalization. For practical statistical analysis, these shapes are typically projected into a tangent Euclidean space where standard multivariate methods can be applied [20]. In drug discovery, this translates to a shape space where each point represents the three-dimensional configuration of a molecule, with distances between points corresponding to their shape dissimilarity.
Molecular Representation Methods
Effective shape-based screening requires appropriate molecular representations that capture critical three-dimensional features:
- Gaussian Molecular Descriptions: Represent atoms as Gaussian functions, enabling rapid shape and electrostatic potential comparisons [42] [40].
- Molecular Surfaces: Define the solvent-accessible surface, which directly interacts with biological targets [42].
- Grid-Based Representations: Discretize molecular properties into three-dimensional grids for computational processing [40].
- Field-Based Similarity: Compare electrostatic potential fields and directional hydrogen-bonding preferences [42].
Table 1: Molecular Representation Methods in Shape-Based Screening
| Representation Type | Key Features | Applications | Limitations |
|---|---|---|---|
| Gaussian Molecular Description | Smooth atomic representation; Fast similarity calculations | High-throughput shape screening [40] | May oversimplify complex molecular features |
| Molecular Surface Shape | Directly models binding interface; Physically meaningful | Pose prediction; Binding site analysis [42] | Computationally intensive for large libraries |
| Electrostatic Field | Captures charge distribution; Incorporates chemical features | Selectivity screening; Specificity prediction [42] | Sensitive to conformational changes |
| Grid-Based Representation | Discrete spatial sampling; Compatible with GPU acceleration | Ultralarge library screening [40] | Resolution-dependent performance |
Computational Methodologies for 3D Shape Similarity
Core Algorithms and Approaches
Shape-Only Similarity Methods
The Rapid Overlay of Chemical Structures (ROCS) algorithm and its GPU-accelerated counterpart FastROCS represent widely adopted approaches for 3D shape similarity searching. These methods employ a Gaussian description of molecular shape that enables rapid overlay and scoring of molecular alignments [40]. The fundamental operation involves maximizing the volume overlap between two molecules through rotational and translational optimization, producing a Tanimoto-like shape similarity score ranging from 0 (no overlap) to 1 (perfect overlap).
The underlying algorithm performs molecular alignment through 3D rotation and translation, optimizing the overlap volume defined by:
[ \text{ShapeTanimoto} = \frac{\int VA(\mathbf{r}) VB(\mathbf{r}) d\mathbf{r}}{\int VA^2(\mathbf{r}) d\mathbf{r} + \int VB^2(\mathbf{r}) d\mathbf{r} - \int VA(\mathbf{r}) VB(\mathbf{r}) d\mathbf{r}} ]
where (VA) and (VB) represent the volume functions of molecules A and B [40].
Combined Shape and Chemical Feature Similarity
Beyond pure shape comparison, advanced methods incorporate chemical feature matching to improve screening accuracy. The eSim method, for instance, combines electrostatic field comparison with molecular surface-shape analysis and directional hydrogen-bonding preferences [42]. This integrated approach recognizes that successful molecular recognition depends not only on shape complementarity but also on compatible electrostatic interactions and hydrogen-bonding patterns.
This methodology calculates similarity using a weighted approach that considers multiple molecular properties simultaneously, providing a more physiologically relevant similarity measure than shape alone [42].
Hybrid Structure- and Ligand-Based Methods
FastROCS Plus represents an advanced implementation that seamlessly combines ligand-based shape screening with structure-based docking approaches in a single workflow [40]. This hybrid methodology leverages the strengths of both approaches: the scaffold-hopping capability of shape similarity with the precise binding pose prediction of molecular docking.
Diagram 1: 3D Shape Similarity Screening Workflow. This workflow integrates multiple similarity metrics and consensus scoring for hit identification.
Performance Metrics and Benchmarking
Rigorous validation of shape similarity methods typically employs benchmark datasets such as the Directory of Useful Decoys (DUD-E), which contains 102 targets with confirmed active compounds and carefully selected decoy molecules that are chemically similar but physiologically inactive [42]. Performance is evaluated using enrichment metrics that measure the method's ability to prioritize active compounds over decoys.
The standard DUD-E evaluation demonstrated that the eSim method, processing over 60 molecules per second on a single computing core, achieved significant enrichment of active compounds across multiple target classes [42]. Similarly, FastROCS has demonstrated the capability to process millions to hundreds of millions of conformations per second on GPU hardware, enabling ultralarge library screening campaigns [40].
Table 2: Performance Comparison of 3D Similarity Methods
| Method | Throughput | Enrichment Factor | Key Advantages | Supported Platforms |
|---|---|---|---|---|
| eSim | ~60 molecules/second/core (screening mode) [42] | High on DUD-E benchmarks | Combines shape with electrostatic fields; Physically meaningful | Standalone applications |
| FastROCS | Millions of conformations/second/GPU [40] | Validated in prospective studies [40] | Extreme speed; GPU acceleration; Hybrid screening | Orion web interface; VIDA desktop |
| FrankenROCS | Variable (active learning) | Identified submicromolar inhibitors [40] | Active learning integration; Targets specific properties | Custom pipeline implementation |
Experimental Protocols and Implementation
Protocol 1: Structure-Based Virtual Screening with Shape Similarity
Objective: Identify novel chemotypes for a protein target with known active site geometry.
Materials and Methods:
- Query Generation:
- Extract the binding site cavity from a protein-ligand complex structure
- Create a negative image of the binding site volume
- Define chemical feature constraints (hydrogen bond donors/acceptors, hydrophobic regions)
Database Preparation:
- Convert compound library to 3D conformations using tools like OMEGA
- Generate multiple conformers to account for molecular flexibility
- Apply energy minimization and filter for drug-like properties
Screening Process:
- Perform shape similarity search using the negative image as query
- Apply chemical feature constraints to ensure complementary interactions
- Use consensus scoring combining shape and feature matches
Post-processing:
- Cluster results by chemical scaffold to ensure structural diversity
- Apply additional filters (ADMET properties, synthetic accessibility)
- Select top candidates for experimental validation
This protocol was successfully applied in the FrankenROCS pipeline, which integrated FastROCS with active learning to explore the 22-billion-molecule Enamine REAL database, identifying submicromolar inhibitors of the SARS-CoV-2 macrodomain with improved cell permeability [40].
Protocol 2: Ligand-Based Virtual Screening for Scaffold Hopping
Objective: Find structurally diverse compounds with similar biological activity to a known active molecule.
Materials and Methods:
- Query Preparation:
- Select a known active compound as the query molecule
- Generate a biologically relevant conformation (from crystal structure or conformational analysis)
- Define critical chemical features (pharmacophore pattern)
Similarity Search:
- Perform 3D shape similarity search against a compound library
- Use combined shape and feature similarity scoring (e.g., ComboScore in ROCS)
- Set appropriate similarity thresholds based on validation experiments
Result Analysis:
- Analyze hit lists for structural diversity relative to the query
- Validate scaffold hops through binding mode analysis or molecular docking
- Prioritize compounds with optimal physicochemical properties
This approach has proven particularly valuable for circumventing existing patents and optimizing drug-like properties while maintaining biological activity [41]. The method enables identification of novel molecular scaffolds that would be missed by traditional 2D similarity methods.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Computational Tools for 3D Shape Similarity Screening
| Tool/Platform | Type | Primary Function | Application Context |
|---|---|---|---|
| FastROCS [40] | Software Suite | GPU-accelerated shape similarity search | Ultralarge library screening; Lead hopping |
| Orion Modeling Platform [40] | Web Interface | Cloud-based molecular modeling | Accessible screening without local hardware |
| VIDA [40] | Desktop Visualizer | Molecular visualization and analysis | Result interpretation and visualization |
| DUD-E Dataset [42] | Benchmark Database | 102 targets with actives and decoys | Method validation and performance assessment |
| Enamine REAL Library [40] | Compound Database | 22+ billion make-on-demand compounds | Ultralarge virtual screening campaigns |
| CETSA [43] | Experimental Assay | Cellular target engagement validation | Experimental confirmation of computational predictions |
Emerging Trends and Integrative Approaches
Artificial Intelligence and Deep Learning
Recent advances in artificial intelligence are transforming shape-based virtual screening through multitask learning frameworks that simultaneously predict drug-target affinity and generate novel target-aware compounds. The DeepDTAGen model exemplifies this approach, using shared feature representations for both predictive and generative tasks [44]. This model demonstrated superior performance on benchmark datasets (KIBA, Davis, BindingDB), achieving MSE of 0.146, CI of 0.897, and r²m of 0.765 on the KIBA test set [44].
These AI-driven approaches leverage graph neural networks and transformer architectures to capture complex structure-activity relationships, moving beyond traditional predefined molecular descriptors to learned representations that better predict biological activity [41].
Automation and High-Throughput Workflows
Modern drug discovery increasingly relies on automated, integrated workflows that combine computational screening with experimental validation. Platforms such as the eProtein Discovery System enable researchers to move from DNA to purified, active protein in under 48 hours, dramatically accelerating the validation of computational predictions [45]. Similarly, automated 3D cell culture systems like the MO:BOT platform enhance the physiological relevance of screening data by providing more human-predictive models [45].
Diagram 2: Integrated Drug Discovery Pipeline. Modern workflows combine computational and experimental approaches with feedback loops for iterative optimization.
The application of 3D shape similarity in virtual screening and target prediction represents a powerful methodology grounded in the fundamental principles of geometric morphometrics. By quantifying molecular complementarity through shape space analysis and Procrustean alignment techniques, researchers can efficiently navigate vast chemical spaces to identify novel therapeutic candidates. The integration of these approaches with artificial intelligence, automated workflows, and human-relevant biological models promises to further accelerate drug discovery while reducing attrition rates in later development stages.
As the field advances, the convergence of shape-based screening with predictive AI models and high-throughput experimental validation creates a virtuous cycle of innovation. This integrative approach, framed within the rigorous mathematical context of morphometric shape analysis, positions 3D molecular similarity as an indispensable tool in the modern drug discovery arsenal, capable of addressing the complex challenges of therapeutic development in the era of personalized medicine.
The Challenge of Nutritional Status Assessment
Nutritional assessment is fundamental to public health, particularly for vulnerable populations in both clinical and field settings. Traditional nutritional assessment relies on the ABCD methods: Anthropometry, Biochemical/biophysical methods, Clinical methods, and Dietary methods [46]. Among these, anthropometry—the measurement of human body dimensions—provides critical objective data for identifying malnutrition. Arm anthropometry serves as a proxy measure for body composition, assessing muscularity, fat-free mass, and fat mass through measurements including upper arm length, mid-upper arm circumference (MUAC), and triceps skinfold (TSF) [47]. These measurements derive indices like arm muscle area (AMA), arm fat area (AFA), and arm fat index (AFI) for comprehensive nutritional evaluation [47]. While these methods are inexpensive, non-invasive, and suitable for field use, they traditionally require manual measurement, introducing potential observer variability and limiting scalability [47].
Geometric Morphometrics and Shape Space Theory
Current alignment-based methods for classification in geometric morphometrics face a significant limitation: they generally cannot directly classify new individuals that were not part of the original study sample [18]. This creates a practical problem for nutritional assessment from body shape images, as classification rules obtained from a reference sample in shape space cannot be applied to out-of-sample individuals in a straightforward manner [18]. Geometric morphometrics provides a sophisticated approach to quantifying biological form using Cartesian coordinates of anatomical landmarks, offering powerful statistical analysis of shape variation while preserving geometric information throughout the analytical process.
The fundamental challenge lies in the sample-dependent processing steps required before classification, including alignment through Procrustes analysis and allometric regression [18]. This work addresses this gap by proposing methods for obtaining shape coordinates for new individuals and analyzing how different template configurations affect registration accuracy of out-of-sample raw coordinates [18]. Understanding sample characteristics and collinearity among shape variables proves crucial for optimal classification results when evaluating children's nutritional status using arm shape analysis from photographs [18]. This approach aligns with initiatives like the SAM Photo Diagnosis App Program, which aims to develop offline smartphone tools capable of updating training samples across different nutritional screening campaigns [18].
Methodology and Technical Implementation
Data Acquisition and Preprocessing
The foundation of accurate automated assessment lies in robust data acquisition. A structured approach using 3D depth-sensing cameras enables precise capture of arm morphology. Research demonstrates that a commercially available ASUS Xtion Pro 3D depth-sensing camera, combined with specialized software, can generate triangulated 2D manifolds of the arm surface exported as STL-files containing vertices and connectivity of 3D points [48]. A standardized scanning protocol developed for clinical use requires approximately 20-30 seconds per scan, utilizing an inexpensive rig (under 500 GBP) consisting of a camera tripod, ball joint mount, and customized camera mount [48].
During scanning, patients should be seated on a stool with their arm stretched out horizontally at the same height as the camera ball joint. The camera rotates 360° around the arm to capture comprehensive shape data [48]. This raw 3D data then undergoes crucial preprocessing:
- Cleaning: Removal of unwanted objects and noise from scans
- Rotation and Cropping: Standardization of scan orientation and focus area
- Patching: Completion of any missing surface data
- Surface Triangulation: Generation of continuous surfaces from point clouds [48]
For large-scale implementation, researchers have proposed IoT-enabled anthropometric data acquisition systems that enhance real-time monitoring and scalability [49].
Deep Learning Framework for Shape Analysis
The DeepSSM framework provides a sophisticated approach for extracting low-dimensional shape representations directly from 3D images, requiring minimal parameter tuning or manual intervention [50]. This convolutional neural network simultaneously localizes the biological structure of interest, establishes correspondences, and projects these points onto a low-dimensional shape representation in the form of PCA loadings within a point distribution model [50].
Table 1: DeepSSM Network Architecture Specifications
| Layer Type | Number | Activation Function | Additional Features |
|---|---|---|---|
| Convolutional | 5 | Parametric ReLU | Batch Normalization |
| Fully Connected | 2 | - | Xavier Initialization |
To address the challenge of limited training data, a novel augmentation procedure uses existing correspondences on a relatively small set of processed images (typically 40-50 samples) with shape statistics to create plausible training samples with known shape parameters [50]. This leverages limited CT/MRI scans into thousands of training images needed for deep neural network training through a process of statistical shape model generation and thin-plate spline warping [50].
Shape Classification and Nutritional Status Prediction
For nutritional classification, advanced deep learning frameworks enhanced with Multi-Head Attention demonstrate significant promise. Research shows that CNN-MHA architectures achieve superior performance (99.08% accuracy) compared to LSTM-MHA (98.91%) on structured anthropometric tabular data, confirming that spatial modeling outperforms sequential dependency approaches for this data type [49]. Integration with Explainable AI techniques, particularly SHapley Additive exPlanations, provides model transparency by identifying the most influential predictors aligned with WHO standards [49].
The geometric morphometrics approach specifically addresses the critical challenge of classifying new individuals not included in the original sample [18]. By developing methods to obtain shape coordinates for out-of-sample individuals and analyzing the effect of different template configurations for registration, this approach enables practical application in nutritional screening campaigns where each new subject constitutes an "out-of-sample" case [18].
Experimental Validation and Performance
Methodological Protocols
Validation of automated arm shape assessment requires rigorous experimental protocols. In studies investigating lymphoedematous arms, researchers recruited 24 patients with mild unilateral lymphoedema, comparing affected and healthy arms using shape-related metrics like circumference and circularity [48]. The protocol involved:
- Patient Positioning: Seated position with arm horizontally extended
- Scanning Procedure: 360° rotation around arm in 20-30 seconds
- Comparison Method: Tape measurements at 4cm intervals for validation
- Analysis Framework: Shape metrics calculation and swelling map derivation [48]
For nutritional assessment, the geometric morphometrics approach requires specific consideration of sample characteristics and collinearity among shape variables [18]. The experimental workflow involves:
- Template Selection: Choosing optimal template configurations for out-of-sample registration
- Procrustes Analysis: Superimposing shapes to remove non-shape variation
- Allometric Correction: Accounting for size-related shape changes
- Classification Modeling: Applying machine learning algorithms to shape variables
Performance Metrics and Benchmarking
Recent machine learning approaches for nutritional assessment demonstrate impressive performance metrics. The XGBoost algorithm has shown particular promise in malnutrition prediction, achieving an accuracy of 0.90 with precision of 0.92, recall of 0.92, F1 score of 0.92, and AUC-ROC of 0.98 in development phases [51]. External validation confirms robust performance with accuracy of 0.75 and AUC-ROC of 0.88 [51].
Table 2: Performance Comparison of Nutritional Assessment Models
| Model Type | Accuracy | Precision | Recall | AUC-ROC | Application Context |
|---|---|---|---|---|---|
| CNN-MHA | 99.08% | - | - | - | Anthropometric Data |
| XGBoost | 90.00% | 92.00% | 92.00% | 98.00% | ICU Malnutrition |
| LSTM-MHA | 98.91% | - | - | - | Anthropometric Data |
| XGBoost (External) | 75.00% | 79.00% | 75.00% | 88.00% | ICU Malnutrition |
Deep learning frameworks for shape analysis demonstrate efficient training characteristics, with empirical observations showing error stabilization after 50 epochs within a range of 1.9-2.5, typically reaching optimal performance after 60 epochs of training [50].
Implementation and Clinical Translation
Research Reagents and Technical Solutions
Table 3: Essential Research Reagents and Technical Solutions
| Item | Function | Specifications | Application Note |
|---|---|---|---|
| ASUS Xtion Pro 3D | Depth sensing camera | Infrared radiation detection, 20-30s scan time | Captures triangulated 2D manifolds as STL files [48] |
| ShapeWorks Software | Correspondence point optimization | Open-source platform | Requires extensive preprocessing of 3D images [50] |
| Salter Scale | Weight measurement | Spring balance, 0.1kg precision | For children under two years; can be improvised with basin [46] |
| Non-stretchable Insertion Tape | MUAC measurement | Millimeter graduation, color-coded cutoffs | Varies by population (infants, children, adults) [47] |
| Sliding Board | Length measurement | Wooden board, millimeter precision | For children under two years; requires assistant [46] |
Clinical Integration and Workflow
The transition from research validation to clinical implementation requires careful workflow design. Automated nutritional assessment systems must integrate seamlessly with existing clinical practices while providing tangible improvements in efficiency and accuracy. The SAM Photo Diagnosis App Program exemplifies this approach, aiming to develop an offline smartphone tool that enables updates of training samples across different nutritional screening campaigns [18].
For successful implementation, automated systems must address several practical considerations:
- Field Conditions: Equipment must be portable, inexpensive, and operable with minimal training
- Data Processing: Algorithms should function offline or with limited connectivity
- Interpretability: Results must be clinically actionable with transparent reasoning
- Integration: Systems should complement rather than replace clinician judgment
The arm anthropometry method offers particular advantages in resource-limited settings, requiring large participant numbers at low cost with minimal burden to participants or researchers [47]. However, limitations include the need for population-specific cutoff values and potential observer variability in measurement technique [47].
Automated nutritional status assessment from arm shape represents a significant advancement in geometric morphometrics, addressing the critical challenge of classifying out-of-sample individuals through sophisticated shape space modeling. The integration of 3D imaging technologies with deep learning frameworks enables accurate, scalable nutritional assessment that transcends the limitations of traditional manual methods. As research in this field evolves, future directions should explore multi-modal data integration, enhanced generalization across diverse populations, and refined visualization techniques for clinical interpretation. By bridging the gap between high-accuracy artificial intelligence and clinical transparency, these automated assessment systems offer promising tools for public health interventions, clinical monitoring, and nutritional research across diverse global contexts.
High-throughput phenotyping represents a paradigm shift in biological research, enabling the rapid, accurate, and large-scale collection of morphological data. At its core lies the concept of shape space—a mathematical construct in which each organism or structure is represented as a single point whose coordinates are defined by its morphological attributes [52]. The transition from traditional manual morphometrics to automated approaches has fundamentally transformed our ability to navigate and classify within this shape space. Traditional methods relying on manual caliper measurements are plagued by limitations in throughput, consistency, and the ability to capture complex geometric shapes. These constraints are overcome by geometric morphometrics, a method that allows the determination of differences and similarities between species of biological shapes through statistical analysis of landmark coordinates [53].
The analytical foundation of these automated methods is Procrustes analysis, a statistical technique that normalizes raw landmark coordinates by removing differences in position, scale, and orientation, allowing for pure shape comparison [54] [53]. This process facilitates the creation of a shared shape space where biological similarity can be quantitatively assessed. Subsequent Principal Component Analysis (PCA) of these Procrustes coordinates then identifies the major axes of shape variation within a sample, effectively mapping the most important dimensions of the shape space [53]. For example, in a study of astragalus bones across bovine, ovine, and caprine species, the first four principal components collectively explained 61.84% of the total shape variation, providing a reduced-dimensionality framework for effective taxonomic classification [53].
The High-Throughput Phenotyping Toolbox
The growing demand for automated morphometric analysis has spurred the development of specialized software tools that combine computer vision with machine learning to streamline the landmarking process. These tools vary in their technical approach, accessibility, and specific applications, but share the common goal of enabling efficient navigation through shape space.
Table 1: High-Throughput Phenotyping Tools for Shape Analysis
| Tool Name | Primary Methodology | Key Features | Accessibility | Documented Accuracy |
|---|---|---|---|---|
| HusMorph | Machine learning-based landmark prediction | GUI for non-experts, automated parameter optimization, scale bar detection | Standalone executable (Windows/Mac), no coding required | ~99.5% compared to manual measurements [55] |
| SPACe | Unsupervised shape and appearance modeling | Generative modeling, handles missing data, privacy-preserving latent variables | Programming expertise required, implemented in research frameworks | Competitive classification accuracy on MNIST with limited training examples [52] |
| MorphoJ | Traditional geometric morphometrics | Procrustes analysis, PCA, discriminant function analysis | Desktop application, menu-driven interface | Validated for distinguishing invasive vs. native moth species [56] |
HusMorph: Accessible Machine Learning for Non-Experts
HusMorph exemplifies the trend toward democratizing high-throughput phenotyping through user-friendly interfaces. This application packages sophisticated machine learning capabilities into an accessible graphical user interface (GUI), eliminating the need for programming expertise [55]. The system is designed as an all-in-one package that guides users through the complete workflow: from manual landmark placement on a training set of images, through automated model training, to applying the trained model to new images with predictive landmarking [57].
A key innovation in HusMorph is its automated hyperparameter optimization using the Optuna library, which randomly searches for the best-performing parameters within defined ranges [55]. This eliminates what is traditionally a major technical barrier for non-expert users—manually tuning complex machine learning parameters. The application employs dlib's machine learning library with a standard CPU setup, making it compatible with conventional desktop computers and laptops, though very high-resolution images may require hardware considerations [55]. For biological research applications, an additional valuable feature is the automated scale bar detection, which converts pixel measurements to metric units, enabling direct biological interpretation of results [57].
SPACe: Unsupervised Shape and Appearance Modeling
In contrast to HusMorph's supervised approach, the SPACe (Shape and Appearance Modeling) algorithm represents a more advanced unsupervised framework for automatically learning shape and appearance models from medical and biological images without manual annotations [52]. This method builds upon Principal Geodesic Analysis (PGA) within the diffeomorphic setting, creating a generative model that captures both shape variability through deformable transformations and appearance variability through signal adaptations.
The mathematical foundation of SPACe involves modeling shape using Large-Deformation Diffeomorphic Metric Mapping (LDDMM), which ensures that deformations between shapes are smooth, invertible, and one-to-one [52]. Appearance is modeled separately as a linear combination of basis functions: an = μ + Wazn, where μ is a mean image, Wa contains appearance basis functions, and zn represents latent variables [52]. These latent variables serve as compact representations within the shape space and can be used as features for privacy-preserving data mining applications—a particularly valuable attribute for multi-site medical studies where patient confidentiality is paramount.
Experimental Protocols and Methodologies
Workflow for Automated Landmarking with HusMorph
The implementation of HusMorph follows a structured protocol that balances automation with expert oversight:
Image Acquisition and Preparation: Images should be captured with standardized rotation, flipping, and scaling against a homogeneous background distinct from the subject. Recommended resolution is ≤2 megapixels to balance detail and computational efficiency [55].
Training Set Creation: A minimum of 100 images with manually placed landmarks by a domain expert establishes the ground truth dataset. The number of landmarks can be customized based on biological relevance [55].
Model Training: The application automatically splits the dataset and performs 5-fold cross-validation while optimizing nine key parameters via the Optuna library. This process is computationally intensive, potentially requiring 1-2 days on modern laptops [55].
Prediction and Validation: The trained model predicts landmarks on new images, with results exportable in CSV format. Visual confirmation is recommended to ensure biological plausibility [57].
Diagram 1: HusMorph automated landmarking workflow
Validation Protocol for Morphometric Applications
Rigorous validation is essential when applying high-throughput phenotyping to classification problems. A study on Chrysodeixis moths exemplifies a robust validation protocol:
Species Validation: Initial specimen identification through male genitalia dissection or real-time PCR testing establishes ground truth [56].
Wing Preparation: Well-preserved right forewings are cleaned and photographed under a digital microscope.
Landmark Annotation: Seven venation landmarks are annotated on wing images, capturing essential shape information while addressing challenges with trap-collected specimens [56].
Data Analysis: Landmark coordinates are analyzed in MorphoJ software, employing Procrustes analysis followed by discriminant function analysis to validate species distinctions [56].
This protocol successfully validated the distinction between invasive C. chalcites and native C. includens, demonstrating the utility of geometric morphometrics for pest identification in survey programs [56].
Shape Descriptor Evaluation Framework
For researchers implementing custom shape analysis pipelines, comparative evaluation of shape descriptors follows this methodological framework:
Contour Extraction and Preprocessing: Cell contours or biological outlines are extracted using deep learning models (CNNs or transformers), resampled to 100 points, and aligned via Procrustes registration [54].
Feature Extraction: Multiple shape descriptors are extracted, including:
- Elliptical Fourier Descriptors: Represent contours using Fourier sine and cosine functions [54]
- Principal Component Analysis: Concatenated coordinates compressed into principal components [54]
- Scalar Features: 23 traditional measurements including circularity, solidity, and eccentricity [54]
- Curvature Features: Rate of change of tangent vector direction along contours [54]
Classification and Evaluation: XGBoost classifier with 5-fold cross-validation assesses performance, with PCA-based approaches demonstrating 99.0% accuracy in synthetic datasets [54].
Research Reagent Solutions for Morphometric Studies
Table 2: Essential Research Reagents and Tools for High-Throughput Phenotyping
| Item Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| Imaging Equipment | Canon 600D with 18×55 lens [53] | Digital capture of morphological specimens | Standardized magnification and lighting critical |
| Specimen Staining | Nissl staining method [54] | Highlights cell body morphology in tissue sections | Essential for neural tissue morphometrics |
| Shape Analysis Software | TpsDig2, TpsUtil [53] | Digitization of landmark coordinates | Establishes homologous landmark sets across specimens |
| Statistical Morphometrics | MorphoJ software [56] [53] | Procrustes analysis, PCA, discriminant functions | Industry standard for geometric morphometrics |
| Machine Learning Libraries | dlib, OpenCV [55] | Core ML algorithms for landmark prediction | HusMorph implementation dependencies |
| Hyperparameter Optimization | Optuna library [55] | Automated parameter tuning for ML models | Eliminates manual optimization requirement |
Advanced Applications and Implementation Considerations
Biological and Biomedical Applications
The implementation of high-throughput phenotyping tools has enabled significant advances across biological disciplines:
Taxonomic Classification: Geometric morphometrics of astragalus bones successfully differentiated bovine, ovis, and capra species with 100% separation between ovis and bovine, and 97.2% separation for capra samples in cross-validation [53]. The analysis revealed significant shape variations at landmarks LM3, LM4, LM8, LM9, LM10, and LM11 between bovine and capras, concentrated primarily on the medial surface of the bone [53].
Invasive Species Monitoring: Wing geometric morphometrics distinguished invasive Chrysodeixis chalcites from native C. includens moths, providing a valuable tool for biosecurity and pest management programs [56]. This approach addressed the limitations of traditional identification methods that require time-consuming male genitalia dissection or DNA analysis.
Biomedical Research: The SPACe algorithm has been applied to a dataset of over 1,900 segmented T1-weighted MR images, demonstrating the potential of shape and appearance modeling for classifying individuals into patient groups in neuroimaging studies [52].
Computational Framework for Shape and Appearance Modeling
The SPACe algorithm employs a sophisticated generative framework that simultaneously learns shape and appearance variability:
Diagram 2: SPACe generative model for shape and appearance
This framework implements a probabilistic approach where the likelihood is summarized as p(fn|zn,μ,Wa,Wv) = p(fn|an(ψn)), with diffeomorphic deformations (ψn) computed from velocity fields (vn) via geodesic shooting [52]. The model can handle missing data—a common challenge in biomedical imaging—and generates latent variables that serve as compact representations for pattern recognition and classification tasks.
Implementation Best Practices
Successful implementation of high-throughput phenotyping requires attention to several critical factors:
Image Standardization: Consistent orientation, background, and scaling dramatically improve model performance. Homogeneous backgrounds with colors distinct from the subject facilitate more accurate landmark prediction [55].
Computational Resources: Model training is computationally intensive, potentially requiring 1-2 days on modern laptops. Dedicated workstations or scheduling for extended computations may be necessary for large datasets [55].
Dataset Size Requirements: While optimal training set size depends on complexity, a minimum of 100 images is recommended. Larger datasets generally improve accuracy, with diminishing returns beyond certain size thresholds [55].
Validation Strategy: Independent validation against manual measurements or established identification methods is crucial. HusMorph achieved ~99.5% accuracy compared to manual measurements on zebrafish standard length [55], while geometric morphometrics approaches showed 93% precision on synthetic fire patterns and 83% on real-world data in non-biological applications [58].
High-throughput phenotyping tools like HusMorph and SPACe represent a transformative advancement in morphometric research, enabling efficient navigation through shape space for classification and analysis. While HusMorph lowers the barrier to entry with its user-friendly interface and automated machine learning, SPACe offers a more sophisticated framework for unsupervised shape and appearance modeling. The integration of these tools with established geometric morphometrics protocols creates a powerful ecosystem for quantitative shape analysis across biological, biomedical, and paleontological disciplines. As these technologies continue to evolve, they promise to further democratize access to advanced shape analysis while increasing the scale, complexity, and reproducibility of morphometric research.
Navigating Challenges: Error Management and Workflow Optimization
In geometric morphometrics (GM), the quantification of phenotypic variation is foundational to addressing a wide range of biological questions. The reliability of these quantitative investigations, however, critically depends on recognizing, quantifying, and mitigating measurement error (ME). When morphological variation is subtle, as is often the case in taxonomic studies, growth analyses, or medical applications, the signal of interest can be easily obscured or falsely generated by biases introduced during data acquisition [59]. These biases are broadly categorized into intra-operator error (variation introduced by a single operator across repeated measurements) and inter-operator error (systematic differences between multiple operators) [60]. In the context of shape space and classification, these errors introduce noise or systematic distortion into the morphospace, potentially leading to misclassification of specimens or incorrect inferences about shape differences and their causes. As morphometrics increasingly moves toward pooling datasets from multiple sources and operators to increase sample size and statistical power, understanding these error sources becomes not merely a methodological formality but a fundamental prerequisite for valid scientific conclusions [59].
Theoretical Framework: Error in the Context of Shape Space
The Procrustes Paradigm and Error Propagation
Geometric morphometric analyses typically begin with Procrustes superimposition, a process that aligns landmark configurations by removing differences due to location, scale, and orientation, leaving only the variation in shape [20]. This process projects raw landmark coordinates into a non-linear shape space, which is then approximated by a linear tangent space for statistical analysis. Within this framework, measurement error does not simply add random noise; it can systematically distort the structure of shape space itself.
When landmarks are digitized with error, this error is carried through the Procrustes alignment. The Generalized Procrustes Analysis (GPA) minimizes the sum of squared distances between corresponding landmarks across specimens, meaning that misplacement of a landmark by one operator can influence the alignment of all other specimens in the dataset [61]. This is particularly critical for classification tasks, where the goal is to define regions of shape space corresponding to different groups (e.g., species, nutritional states, or disease subtypes). Intra- and inter-operator biases can cause specimens to be plotted in incorrect locations within this space, leading to overlap between distinct groups or artificial separation within a homogeneous group, thereby compromising the accuracy of any subsequent classifier [17].
The Problem of "Big Data" and Pooled Datasets
The emerging era of "big data" in morphometrics, involving large-scale collaborative studies and the merging of datasets from different sources, amplifies the risk of inter-operator bias [59] [60]. Pooling data from multiple operators can introduce an excess of variation that masks the true biological signal. A study on human head MRIs demonstrated that inter-operator differences could account for over 30% of the total shape variation in a sample, an effect so substantial that it dominated the main pattern of biological variation, such as sex differences, across hundreds of individuals [60]. This finding underscores a critical point: even with precise landmark definitions, the effect of error on shape can be disproportionately large and must be quantified relative to the total sample variance within the specific methodological context.
Quantifying Intra- and Inter-Operator Bias
A rigorous assessment of measurement error is a necessary step in any morphometric study, especially those intending to pool data or detect subtle phenotypic signals.
Analytical Workflow for Error Assessment
A robust workflow for evaluating whether morphometric datasets can be pooled involves a structured comparison of intra- and inter-operator errors [59]. The following diagram illustrates this process:
Quantitative Benchmarks from Empirical Studies
Empirical studies across different biological disciplines provide critical benchmarks for the magnitude of inter-operator bias. The following table synthesizes key quantitative findings:
Table 1: Quantitative Impact of Inter-Operator Bias in Morphometric Studies
| Biological System | Landmark Type | Reported Impact of Inter-Operator Bias | Reference |
|---|---|---|---|
| Human Head MRI | 3D hard- and soft-tissue landmarks | Accounted for >30% of total sample shape variation, dominating biological signals like sex differences. | [60] |
| Papionin Crania | 3D anatomical landmarks | Variation due to inter-operator differences was substantial, affecting taxonomic classification. | [61] |
| Macropodoid Marsupials | 3D anatomical landmarks | Inter-operator variability accounted for ~8-12% of the total sum of squares for shape. | [60] |
| Sus scrofa Teeth | 2D landmarks & semi-landmarks | Systematic inter-operator bias identified as a major risk for invalidating pooled datasets. | [59] |
These figures demonstrate that the impact of bias is highly context-dependent, varying with the anatomical structures, landmark types, and the experience of the operators. Therefore, a one-size-fits-all threshold for acceptable error does not exist; it must be evaluated relative to the biological effect size under investigation.
Experimental Protocols for Error Quantification
Protocol 1: Replicated Digitization for Variance Component Analysis
This foundational protocol is designed to formally partition variance into its biological and error components.
- Objective: To quantify the proportion of total shape variance attributable to intra- and inter-operator error.
- Materials: A subset of specimens (e.g., 5-10) from the main study sample.
- Procedure:
- Replication: Multiple operators (e.g., 3 or more) digitize the entire landmark set for each specimen in the subset multiple times (e.g., 3 repeated trials). The order of specimens should be randomized between trials to avoid drift.
- Data Structure: The resulting data is structured with multiple observations per individual, per operator.
- Statistical Analysis: A Procrustes ANOVA is performed, which is a specialized form of ANOVA designed for shape data. This model decomposes the total shape variance into components:
- Interpretation: The key outcome is the ratio of variance components. If the variance explained by
OperatorandResidualis a significant proportion (e.g., >10-20%) of theIndividualvariance, the risk of bias is high. This protocol was successfully applied in a study of human os coxae, where it helped determine the optimal coordinate point density and assess the impact of missing data [20].
Protocol 2: Evaluating the Impact on Classification Accuracy
This protocol directly tests how measurement error affects the primary goal of many studies: accurate classification.
- Objective: To determine if inter-operator bias leads to misclassification of specimens in a morphospace.
- Materials: A dataset with known a priori groups (e.g., species, disease status).
- Procedure:
- Baseline Model: A classifier (e.g., Linear Discriminant Analysis) is built using a training set digitized by a single, reference operator. Its performance is evaluated on a test set from the same operator.
- Bias Test: The same classifier is used to predict the group membership of specimens digitized by a different operator.
- Comparison: Classification accuracy (e.g., percentage correctly identified) is compared between the baseline and bias test scenarios. A significant drop in accuracy indicates that inter-operator bias is large enough to compromise the classification rule [17].
- Interpretation: This approach is highly relevant for applied contexts like nutritional assessment, where a classification rule derived from one dataset is applied to new, out-of-sample individuals. If the new data is collected by different operators, bias can render the rule ineffective without proper calibration [17].
The Scientist's Toolkit: Essential Reagents and Materials
The following table details key resources and methodological considerations for designing robust error-assessment experiments.
Table 2: Research Reagent Solutions for Error Assessment in Morphometrics
| Item / Concept | Function / Role in Error Management | Example / Specification |
|---|---|---|
| 3D Structured-light Scanner | Creates high-resolution 3D models of specimens, serving as the primary data source for digitization, reducing error from specimen handling. | Artec Eva scanner [20]. |
| Digitization Software | Platform for placing landmarks and semi-landmarks on 2D images or 3D models. Standardization is key. | tpsDig2, Viewbox 4 [59] [20]. |
| Semi-landmarks | Points placed along curves or surfaces to capture outline shape. Their number and sliding algorithm can be a major source of error and data inflation. | Requires careful protocol for spacing and sliding (e.g., minimum bending energy) [59] [61]. |
| Procrustes ANOVA | Statistical framework for partitioning variance in shape data into biological signal, inter-operator bias, and intra-operator error. | Implemented in software like geomorph (R) [59] [60]. |
| Template Configuration | A standardized set of landmarks and semi-landmarks. Using a common, well-defined template is critical for reducing inter-operator bias. | e.g., A predefined template for the human os coxae [20] [17]. |
Mitigation Strategies and Best Practices
Optimizing Data Acquisition and Analysis
Minimizing error requires a proactive approach throughout the research pipeline, from study design to data analysis.
- Operator Training and Calibration: Implement a training period where all operators digitize a common set of specimens. Discuss discrepancies to converge on a consistent interpretation of landmark definitions [60].
- Landmark Selection and Definition: Prioritize Type I landmarks (defined by discrete anatomical loci, e.g., sutures) over Type II (mathematically defined, e.g., extremal points) or Type III (constructed, e.g., midpoints), as they are generally more reliable [60]. For semi-landmarks, perform pilot studies to determine the optimal density that captures morphology without introducing redundant variables and over-inflating dimensionality [59] [20].
- Protocol Validation with Pilot Data: Before commencing a full-scale study, conduct a pilot study using the replicated digitization protocol (Section 4.1). This allows for the a priori assessment of whether the chosen morphometric protocol has sufficient power to detect the effect of interest above the noise level of the ME [59].
- Use of Supervised Machine Learning Classifiers: Recent critiques suggest that the standard practice of Principal Component Analysis (PCA) followed by visual inspection of scatterplots is highly susceptible to producing artifactual patterns and is less reliable for classification than supervised machine learning methods. Classifiers like those implemented in the
MORPHIXpackage can provide more accurate classification and better detection of new taxa or groups, reducing the subjective interpretation of biased data [61].
A Framework for Data Pooling
The decision to pool datasets from multiple operators should be evidence-based. The following diagram outlines a logical decision-making process:
Intra- and inter-operator biases are not merely nuisances in morphometric research; they are fundamental parameters that must be quantified and reported. In the context of shape space and classification, these biases can distort the very structure of the morphospace, leading to incorrect classifications and flawed biological inferences. As the field moves toward larger, pooled datasets and more automated classification tools, a rigorous, statistically grounded approach to error assessment becomes indispensable. The protocols and mitigation strategies outlined here provide a roadmap for researchers to ensure that their conclusions about phenotypic variation and classification are built upon a foundation of reliable and reproducible data.
In the field of morphometrics, where quantitative analysis of shape is paramount, researchers routinely build classification systems to categorize specimens based on their geometric properties. These may be used for applications ranging from identifying species from fossil records to assessing nutritional status in children [17] [29]. A fundamental challenge emerges when a classification rule, developed from a carefully studied reference sample, must be applied to a new individual that was not part of the original study. This is known as the out-of-sample problem. In traditional morphometric approaches using linear measurements, applying a established discriminant function to a new specimen is straightforward, as the same measurements are simply taken anew [17]. However, in geometric morphometrics (GM), classifiers are typically constructed not from raw coordinates but from transformed data that has undergone a sample-dependent process, such as Generalized Procrustes Analysis (GPA), which aligns all specimens in a dataset into a common shape space [17] [62].
The core of the out-of-sample problem is that the aligned coordinates for a new individual cannot be obtained without including them in a new, global alignment with the original sample, which is often impractical or violates the principles of proper model validation [17]. This whitepaper details the theoretical underpinnings of this problem and outlines robust, practical strategies for classifying new individuals within the context of shape space and morphometrics research. Understanding and overcoming this hurdle is critical for deploying reliable, real-world classification systems in fields like paleontology, drug development, and clinical diagnostics [17] [29].
Theoretical Framework: Shape Space and the Out-of-Sample Hurdle
Defining Shape Space and Pre-Shape
In statistical shape analysis, the shape of an object is all the geometric information that remains after discounting the effects of translation, scale, and rotation [62]. The process of extracting this information leads to the concept of a shape space. The journey to this space begins with the pre-shape, which is the configuration of landmarks after centering (removing location) and scaling to unit size [62]. The pre-shape sphere is the intermediate stage before the final removal of rotation to align configurations.
- Landmark Configuration: An object is represented by a ( k \times m ) matrix of coordinates, where ( k ) is the number of landmarks and ( m ) is the dimensionality (e.g., ( m=2 ) for planar shapes) [62].
- Generalized Procrustes Analysis (GPA): This is the standard method for aligning a sample of specimens into a common shape space. GPA iteratively translates, scales, and rotates configurations to minimize the total sum of squared distances between corresponding landmarks, resulting in Procrustes shape coordinates [17] [63].
The Statistical Challenge of a Single New Individual
A classifier, such as Linear Discriminant Analysis (LDA), Support Vector Machine (SVM), or a neural network, is trained on the Procrustes coordinates of a reference sample [17] [62] [63]. The out-of-sample problem arises because the Procrustes coordinates for a new specimen are undefined in isolation; they are inherently relational and depend on the entire sample used for the GPA. Conducting a new GPA that includes the new individual is methodologically flawed for a true classification task, as it uses the unknown individual's data to inform the alignment process, potentially biasing the classification and leading to over-optimistic performance estimates [17]. Therefore, a strategy is needed to project the new individual into the existing shape space of the training sample without recalculating that space.
Methodological Strategies for Out-of-Sample Classification
Template Registration Approach
One proposed methodology for evaluating out-of-sample cases involves registration against a template [17]. Instead of performing a full GPA with the entire training set, a single representative configuration from the training sample is selected as a target template.
- Procedure: The raw landmark coordinates of the new individual are aligned to this single template via an ordinary Procrustes analysis (translation, scaling, and rotation). The resulting registered coordinates of the new individual are then treated as being in the shape space of the original training sample and can be fed directly into the pre-trained classifier [17].
- Choosing a Template: The choice of template is critical. It could be the mean shape of the training sample or a configuration from a specific group. The effect of using different template configurations on final classification accuracy is an active area of research, and the optimal choice may depend on the specific characteristics of the sample, such as collinearity among shape variables [17].
Advanced Algorithmic and Ensemble Approaches
Beyond the initial registration, the choice of classification algorithm and the use of ensemble methods can significantly impact the robustness of out-of-sample predictions.
Table 1: Key Classification Algorithms for Morphometric Data
| Algorithm | Type | Key Principle | Applicability to Shape Data |
|---|---|---|---|
| Linear Discriminant Analysis (LDA) | Supervised | Finds linear combinations of variables that best separate classes [64]. | Classic approach; assumes homoscedasticity; can be effective but may struggle with complex, high-dimensional shapes [63]. |
| Support Vector Machine (SVM) | Supervised | Finds an optimal hyperplane (or complex boundary in kernel space) to separate classes [62] [64]. | Highly reliable; can be adapted for complex vectors in shape space; performs well with small sample sizes [62] [63]. |
| Random Forest | Supervised (Ensemble) | Builds many decision trees on random data subsets and aggregates their predictions [64]. | Addresses overfitting; handles complex data sets well; effective for high-dimensional phenotypes [63]. |
| Naive Bayes | Supervised | Applies Bayes' theorem with strong independence assumptions between features [64]. | Useful for probabilistic classification; can perform well on shape data despite its simplifying assumptions [64]. |
| K-Nearest Neighbors (KNN) | Supervised | Classifies a point based on the majority class among its K nearest neighbors in shape space [64]. | Simple, intuitive; directly uses the geometry of the shape space for classification [64]. |
The Power of Ensemble Learning
Ensemble learning, particularly blending or stacking, involves strategically combining multiple individual classifiers (base learners) to create a single, stronger model [63]. A meta-analysis of 33 algorithms across 20 high-dimensional morphometric datasets found that ensemble models achieved the highest performance on average, increasing accuracy by up to 3% over the top base learner [63]. The strength of ensembles lies in their ability to be data-agnostic and their exceptional accuracy across diverse classification tasks, making them a powerful tool for generalizing to new, unseen individuals.
Experimental Protocol for Method Validation
To validate any out-of-sample classification pipeline, a rigorous experimental protocol is essential. The following workflow, implemented in R packages like pheble, provides a standardized framework [63]:
- Data Preprocessing: The full available dataset is superimposed into a common shape space via GPA [63].
- Training-Test Split: The data is split into a training set and a holdout test set. Crucially, the GPA is performed only on the training set. The test set is later projected into the training set's shape space using a template registration method to simulate out-of-sample individuals.
- Model Training: Multiple classifiers (e.g., LDA, SVM, Random Forest) are trained on the Procrustes coordinates of the training set.
- Ensemble Construction: Predictions from the base learners are combined using a blending algorithm (e.g., a generalized linear model or another classifier) on a validation set.
- Out-of-Sample Testing: The final ensemble model and all base learners are evaluated on the holdout test set, which was aligned to the training set's template, providing a realistic measure of performance on unseen data.
Diagram 1: Workflow for out-of-sample classification protocol.
A Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for Morphometric Classification
| Item / Reagent | Function / Explanation |
|---|---|
| Homologous Landmarks | Anatomically corresponding points defined across all specimens; the fundamental data points for shape analysis [62] [63]. |
| Semilandmarks | Points defined along curves and surfaces to capture outline geometry; crucial for analyzing shapes lacking sufficient discrete landmarks [17]. |
| Procrustes Shape Coordinates | The aligned coordinates after GPA; the primary variables used for building classifiers in the shared shape space of the training sample [17] [62]. |
| Template Configuration | A single landmark configuration (e.g., the mean shape) from the training sample; used as a target for registering new, out-of-sample individuals [17]. |
| High-Dimensional Phenotypic Datasets | Large collections of shape data; used for training robust machine learning models and testing their performance across various conditions [63]. |
Ensemble Learning Framework (e.g., R package pheble) |
A software tool that streamlines the process of preprocessing data, training multiple models, and constructing ensemble classifiers for high-dimensional data [63]. |
The out-of-sample problem represents a significant methodological challenge in applied morphometrics, but it is not insurmountable. A robust solution involves a two-pronged approach: a geometric strategy for placing new individuals into an established shape space, such as registration via a carefully chosen template, and a statistical learning strategy for maximizing classification accuracy, with ensemble methods currently standing out as the most consistently high-performing option [17] [63]. As machine learning, particularly deep learning, continues to permeate fields like paleontology and biomedical research, the adherence to rigorous validation protocols that properly account for out-of-sample classification will be paramount for developing reliable, automated diagnostic and identification systems [29]. By framing classification within the rigorous context of shape space and adopting these advanced strategies, researchers can ensure their models are both scientifically valid and practically applicable.
In the evolving field of morphometrics research, the digitization of morphological data presents a critical challenge: maximizing the informational yield from data collection without succumbing to the statistical pitfalls of variable inflation. This whitepaper examines the core principles of optimizing digitization efforts within the context of shape space and classification. We provide a structured analysis of the quantitative landscape, detailed experimental protocols from contemporary research, and clear visualization of workflows to guide researchers and drug development professionals in designing robust, scalable morphological studies. The integration of high-dimensional geometric morphometric (GM) data demands a careful balance, as excessive variable inclusion can lead to model overfitting and reduced out-of-sample classification performance, a concern paramount in biomedical applications such as phenotypic drug screening [17].
The shift from traditional linear measurements to landmark-based geometric morphometrics has fundamentally altered how phenotypic variation is quantified. This GM approach captures the geometry of morphological structures, allowing for sophisticated analyses of shape variation and its covariates, such as allometry or nutritional status [17]. However, this power comes with inherent complexity. The process of digitization—converting physical forms into digital landmark data—directly influences the dimensionality of the statistical analysis. Each landmark and semilandmark introduces new variables, potentially leading to a scenario where the number of variables (p) approaches or exceeds the number of specimens (n). This "variable inflation" jeopardizes the stability of statistical models and the generalizability of classification rules, particularly when applied to new, out-of-sample individuals [17]. Understanding this balance is not merely a technical exercise; it is foundational to constructing reliable classifiers for distinguishing pathological phenotypes in drug development or diagnosing malnutrition in global health [17].
The Quantitative Landscape: Data Challenges in Transformation
The broader digital transformation landscape offers critical context for the specific challenges faced in morphometric research. The following tables summarize key statistics on data project success rates and the primary obstacles encountered.
Table 1: Global Data Transformation Success and Failure Rates
| Metric | Statistic | Context/Source |
|---|---|---|
| Digital Transformation Success Rate | 35% | BCG analysis of 850+ companies (2025) [65] |
| Digital Transformation Failure Rate | 70% | Various consulting studies (2025) [65] |
| Big Data Project Failure Rate | 85% | Gartner analysis (2025) [65] |
| System Integration Failure Rate | 84% | Integration research (2025) [65] |
| Data-Driven Fortune 1000 Companies | 37.8% | NewVantage Partners (2025) [65] |
Table 2: Primary Data Quality and Skills Challenges
| Challenge Category | Specific Statistic | Impact/Detail |
|---|---|---|
| Data Quality | 64% cite it as top challenge [65] | Top data integrity challenge |
| 77% rate quality as average or worse [65] | 11-point decline from 2023 | |
| $3.1 trillion annual cost (US businesses) [65] | Historical IBM estimate of poor quality | |
| Skills Gap | 87% of organizations affected [65] | McKinsey research (2025) |
| 90% face IT shortages by 2026 [65] | Projected $5.5 trillion cost | |
| Only 35% receive adequate training [65] | Despite 75% needing reskilling |
Methodological Framework for Morphometric Classification
A central problem in applied geometric morphometrics is the development of classification rules that can be reliably applied to individuals not included in the original training sample. The standard Generalized Procrustes Analysis (GPA) aligns all specimens in a sample simultaneously, a process that cannot be directly performed on a new, single individual. The following workflow delineates a proposed methodology for out-of-sample classification, addressing the core challenge of balancing data quantity (landmarks) with generalizable results [17].
Diagram 1: Out-of-Sample Classification Workflow.
Workflow Logic and Relationships
The process begins with the acquisition of raw landmark data from a reference training sample. This sample must be carefully designed to represent the known variation in the population (e.g., different nutritional statuses, age groups) [17]. The core alignment step, Generalized Procrustes Analysis (GPA), removes differences in position, scale, and orientation to isolate pure shape information [17]. The resulting Procrustes coordinates form the high-dimensional shape variables used to construct a classifier (e.g., Linear Discriminant Analysis). A critical, often overlooked step is the selection of an optimal template configuration from the training set. This template serves as the target for registering new, out-of-sample individuals, allowing their raw coordinates to be placed into the same shape space as the training data without performing a new global GPA. The choice of this template can significantly impact final classification performance and must be investigated as part of the optimization process [17].
Experimental Protocols: A Case Study in Nutritional Morphometrics
The following detailed methodology is adapted from a recent study on classifying children's nutritional status using arm shape analysis, which serves as an exemplary model for managing digitization effort and variable inflation [17].
Sample Collection and Design
- Participant Recruitment: A sample of 410 Senegalese children (206 girls, 204 boys) aged 6-59 months was recruited. The sample was designed with equal proportions of Severe Acute Malnutrition (SAM) and Optimal Nutritional Condition (ONC) participants, balanced for age and sex [17].
- Inclusion/Exclusion Criteria:
- Inclusion: Children aged 6-59 months; classified as ONC (Mid-Upper Arm Circumference (MUAC) 135-155 mm or Weight-for-Height Z-score (WHZ) -1 to +1 SD) or SAM (MUAC < 115 mm or WHZ < -3 SD); consent form signed by legal guardians [17].
- Exclusion: Physical malformations affecting arm anatomy; medical complications other than SAM; marks or scars on the arm that could enable identification [17].
- Ethical Approval: The study was approved by the Senegalese National Ethical Committee for Health Research. Privacy data protection was ensured according to Senegalese and European laws [17].
Data Acquisition and Digitization
- Anthropometric Measurements: Weight, height/length, and MUAC were recorded using calibrated, certified equipment (e.g., SECA 874 scale, portable infantometer). These measurements provided the gold-standard classification for nutritional status [17].
- Image Capture for Morphometrics: High-quality images of the subjects' left arms were captured under standardized conditions. This step represents the primary digitization effort, converting a complex 3D form into a 2D image for subsequent landmark digitization [17].
- Landmark and Semilandmark Digitization: Landmarks (anatomically defined points) and semilandmarks (points along curves) were placed on the arm images. The number and location of these points determine the initial dimensionality of the dataset. This is a key decision point where the balance between data quantity and variable inflation must be struck [17].
Data Processing and Statistical Analysis
- Procrustes Superimposition: Raw landmark coordinates underwent Generalized Procrustes Analysis to remove non-shape variation [17].
- Template Selection for Out-of-Sample Registration: The study investigated the effect of using different template configurations (e.g., mean shape of the entire sample, mean shape of each nutritional group) from the training sample as targets for registering new individuals. This is a crucial step for making the classification rule applicable in real-world settings, such as a smartphone app [17].
- Classifier Construction and Validation: A linear discriminant analysis classifier was built from the Procrustes coordinates. Performance was rigorously tested using leave-one-out cross-validation to provide an honest assessment of classification accuracy before application to out-of-sample data [17].
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key resources and their functions for executing a morphometric study as described in the experimental protocol.
Table 3: Key Research Reagent Solutions for Morphometric Analysis
| Item Name | Function / Application | Specific Example / Note |
|---|---|---|
| Calibrated Anthropometric Tools | Provides gold-standard physiological measurements for validation. | SECA 874 electronic scale (0.1 kg precision); portable infantometer/height board; MUAC tape [17]. |
| Standardized Imaging System | Captures high-resolution, reproducible 2D images of morphological structures. | Smartphone camera with fixed positioning and lighting to minimize non-biological shape variance [17]. |
| Landmark Digitization Software | Enables precise placement of anatomical landmarks and semilandmarks on digital images. | Software used in the SAM Photo Diagnosis App Program for offline analysis [17]. |
| Geometric Morphometrics Software | Performs core statistical shape analysis (GPA, PCA, DFA). | R packages (e.g., geomorph, Morpho); integrated into analysis pipelines for shape variable extraction [17]. |
| Statistical Computing Environment | Platform for building and validating classification models and performing custom analysis. | R or Python with specialized libraries (e.g., urbnthemes for standardized visualization) [66]. |
Visualizing the Classification Challenge in Shape Space
The fundamental challenge of variable inflation and model generalization can be visualized as a pathway where data quality and model complexity interact. The following diagram illustrates the decision points that lead to either robust classification or model failure, connecting the concepts of data quantity, variable inflation, and generalizability.
Diagram 2: The Digitization Optimization Pathway.
Pathway Logic and Relationships
The pathway begins with the acquisition of high-dimensional landmark data. The critical juncture is the choice of analysis strategy. Path A represents an unoptimized approach where all digitized variables are used directly in model building. This often leads to variable inflation, where the number of variables (p) approaches the number of specimens (n), resulting in statistical models that are overly complex and tailored to noise within the training sample. The consequence is an overfit model with poor out-of-sample performance [17] [65]. Path B represents the optimized approach, which incorporates dimensionality reduction (e.g., PCA on Procrustes coordinates) or variable selection. While this path carries a risk of losing subtle but biologically meaningful shape information, its careful implementation—informed by cross-validation—leads to a generalizable model capable of robust classification of new individuals, which is the ultimate goal of digitization in applied morphometrics [17].
Data pooling, the practice of combining datasets from multiple sources into a single repository for analysis, has become increasingly common across scientific disciplines. In morphometrics research, where quantifying phenotypic variation is fundamental to understanding shape space and classification, data pooling offers the potential to significantly enhance analytical power by increasing sample sizes and enabling larger-scale comparative studies. The emergence of specialized repositories such as MorphoSource, MorphoBank, and the Morpho Museum facilitates this data archiving and sharing, allowing researchers to combine datasets from multiple operators, institutions, and studies [59].
However, pooling morphometric datasets never comes without risk [59]. While the benefits include the ability to detect more subtle morphological variation and strengthen statistical inferences, the process introduces substantial methodological challenges that can compromise research validity if not properly addressed. The central challenge lies in distinguishing true biological signals from artificial variation introduced during the data acquisition and pooling process itself. This technical guide provides a comprehensive framework for assessing and mitigating these risks, with particular emphasis on morphometric applications involving shape space analysis and classification.
Risk Assessment in Data Pooling
Primary Risk Categories
The risks associated with data pooling can be categorized into three primary domains, each with specific implications for morphometric research:
Measurement Integrity Risks: In morphometrics, multiple sources of imprecision can compromise measurement integrity, including poorly defined measurements, structure flexibility, operator experience, and environmental conditions [59]. These can be summarized as methodological, instrumental, and personal sources of error. When data are pooled from multiple sources, error likely increases with acquisition workflow complexity, particularly when combining data obtained through different protocols (e.g., direct specimen measurement vs. digitized 2D or 3D models) [59].
Privacy and Re-identification Risks: When pooling datasets, even those previously considered de-identified, the risk that an anticipated recipient can identify an individual in the resulting dataset increases substantially [67]. As more identifying variables become available for each individual through dataset linkage, the group size for most individuals decreases, resulting in higher re-identification risk [67]. This is particularly relevant in biomedical morphometrics involving human subjects.
Analytical Validity Risks: Pooling data can significantly impact statistical outcomes, potentially masking true signals or creating false positives [68]. In pharmacovigilance, for example, pooling adverse event data from spontaneous and solicited sources has been shown to impact disproportionality analyses, potentially leading to both false negatives and false positives [68]. Similar risks apply to morphometric classification analyses, where artificial variation introduced through pooling may distort true shape space relationships.
Quantitative Risk Assessment Framework
A structured approach to risk assessment before pooling morphometric datasets is essential. The following workflow provides a methodological foundation for evaluating potential data compatibility issues:
Table: Key Risk Factors in Morphometric Data Pooling
| Risk Category | Specific Risk Factors | Impact Level | Detection Methods |
|---|---|---|---|
| Operator Effects | Inter-operator bias, Intra-operator variability, Systematic landmark misplacement | High | Procrustes ANOVA, Measurement error analysis [59] |
| Instrument Effects | Device variability (calipers, 3D scanners, cameras), Resolution differences, Calibration inconsistencies | Medium-High | Technical replicates, Cross-validation [59] |
| Protocol Effects | Landmark definition differences, Slide semilandmark protocols, Specimen preparation methods | High | Protocol comparison, Multivariate analysis of variance [59] |
| Data Structure Effects | Variable naming differences, Missing data patterns, Metadata incompatibility | Medium | Data audit, Metadata assessment [69] |
The analytical workflow for assessing pooling viability involves estimating both within-operator and among-operator biases to determine whether morphometric datasets can be validly combined [59]. This requires comparing intra-operator measurement errors (one per operator) with inter-operator error to ensure that pooled variation reflects biological signals rather than methodological artifacts.
Mitigation Strategies and Best Practices
Technical Mitigation Approaches
Implementing robust technical safeguards is essential for minimizing pooling-related risks in morphometric research:
Strong Data Encryption: Ensure all pooled data is encrypted both at rest and during transmission to protect against unauthorized access [70]. This is particularly critical when pooling data across institutions or when working with sensitive biological specimens.
Implementation of Access Control: Limit access to pooled data through role-based access controls (RBAC) to ensure sensitive information is only available to authorized personnel [70] [71]. The principle of least privilege should guide access permission assignments.
Data Anonymization and Minimization: When pooling data, ensure that any personally identifiable information is properly anonymized [70]. For morphometric data, this may involve removing metadata that could lead to specimen or subject re-identification while retaining biologically relevant information.
Regular Audits and Monitoring: Conduct regular security audits and monitor systems to identify vulnerabilities or suspicious activity [70]. In research contexts, this should include periodic reassessment of data quality and consistency within pooled datasets.
Methodological Standards for Morphometric Pooling
Establishing rigorous methodological standards is particularly crucial for morphometric data pooling where measurement consistency directly impacts analytical validity:
Protocol Harmonization: Prior to pooling, standardize morphometric protocols across datasets, including landmark definitions, digitization procedures, and equipment specifications [59]. The choice of morphometric approach (e.g., landmarks, sliding semilandmarks, outline analyses) influences the amount of error, and this should be consistent across pooled datasets [59].
Error Quantification: Implement comprehensive error assessment using replicated measurements to quantify both intra- and inter-operator variability [59]. This evaluation should specifically address whether measurement errors introduced by various users significantly exceed intra-operator variability based on a set of similar objects.
Cluster-Based Pooling Strategy: Adapt the approach used in environmental science where waterbodies were grouped into clusters based on similar cyanobacterial bloom patterns before pooling data [72]. In morphometrics, this could involve clustering datasets by similar morphological characteristics or experimental conditions before pooling.
Effect Modifier Identification: In clinical and regulatory contexts, identifying effect modifiers (EMs) - intrinsic and extrinsic factors that may affect therapeutic outcomes - is crucial for appropriate pooling strategies [73]. Similarly, in morphometrics, identifying factors that modify shape characteristics (e.g., specimen preparation methods, imaging techniques) allows for more informed pooling decisions.
The following workflow illustrates the decision process for determining appropriate pooling strategies in multi-regional trials, which can be adapted for morphometric research:
Experimental Protocol for Assessing Pooling Viability
For morphometric researchers considering data pooling, the following experimental protocol provides a methodological framework for assessing dataset compatibility:
Objective: To evaluate whether morphometric datasets from multiple operators can be validly pooled for shape space analysis without introducing significant methodological artifacts.
Materials:
- Multiple specimens representing the morphological range of interest
- Multiple operators with varying experience levels
- Standardized imaging equipment (e.g., DSLR camera with micro lens, 3D scanner)
- Digitization software (e.g., tpsDig2)
- Calibration tools and standardized measurement protocols
Procedure:
- Standardized Imaging: Capture high-resolution images or 3D models of all specimens using consistent equipment settings, lighting conditions, and orientation protocols.
- Blinded Digitization: Have each operator digitize the complete set of specimens following a predefined landmark protocol, with specimen order randomized to minimize systematic bias.
- Repeated Measurements: Incorporate intra-operator replication by having each operator re-digitize a subset of specimens (minimum 20%) in separate sessions to assess repeatability.
- Data Collection: Compile landmark coordinates or linear measurements from all operators into a structured dataset with appropriate metadata tagging.
Analysis Workflow:
- Procrustes Registration: Perform Generalized Procrustes Analysis to remove non-shape variation (position, orientation, scale).
- Measurement Error Assessment: Conduct Procrustes ANOVA to partition variance components into biological signal, inter-operator error, and intra-operator error.
- Multivariate Comparison: Perform MANOVA on Procrustes coordinates to test for significant operator effects.
- Classification Accuracy: Assess whether operator identity can be predicted from shape data using discriminant analysis.
- Signal-to-Noise Evaluation: Compare magnitude of operator effects to biological effects of interest (e.g., between-species differences).
Table: Essential Research Reagents and Tools for Morphometric Data Pooling
| Item | Function | Implementation Example |
|---|---|---|
| Hierarchical Clustering | Groups similar datasets before pooling | Grouping waterbodies by similar CB patterns before pooling data [72] |
| Procrustes ANOVA | Partitions variance components | Quantifying inter-operator vs. intra-operator error in morphometrics [59] |
| Effect Modifier Identification | Identifies factors influencing outcomes | Determining intrinsic/extrinsic factors affecting drug response before pooling [73] |
| Data Anonymization | Protects privacy in combined datasets | Removing personally identifiable information before pooling [67] |
| Role-Based Access Control | Manages data security | Limiting dataset access to authorized researchers only [70] |
Implementation Framework
Decision Framework for Pooling Viability
Implementing a structured decision framework is essential for determining when data pooling is methodologically appropriate. The following criteria should be evaluated before proceeding with dataset combination:
Measurement Error Thresholds: Pooling is generally justified when inter-operator error variance is less than 50% of the biological variance of interest, based on Procrustes ANOVA results [59]. This ensures that biological signals remain dominant in the pooled dataset.
Sample Size Considerations: Data pooling shows greatest benefits when individual dataset sizes are small (e.g., <100 specimens), with performance gains plateauing near several hundred observations (400-500 samples) [72]. Beyond this point, additional data may contribute less to statistical power while increasing heterogeneity.
Protocol Compatibility: Datasets are suitable for pooling when they share core methodological protocols, including similar landmark schemes, equivalent imaging resolutions, and comparable specimen preparation methods [59] [69]. Significant protocol differences generally preclude valid pooling.
The following diagram illustrates the complete experimental workflow for assessing pooling viability in morphometric research:
Special Considerations for Classification in Morphometrics
When data pooling is conducted specifically for shape space analysis and classification tasks, additional considerations apply:
Feature Selection Optimization: In geometric morphometrics, the inflation of variables through sliding semilandmarks often creates high-dimensional datasets that may not improve classification accuracy [59]. Optimizing the number of variables relative to available observations is essential before pooling.
Batch Effect Correction: When pooling datasets from different sources, implement statistical methods to correct for systematic technical variation (batch effects) that could distort true shape space relationships. This may include ComBat or other normalization approaches commonly used in genomics.
Cross-Validation Strategy: Employ stratified cross-validation that maintains representation from each original dataset in training and test splits to ensure classification models generalize across sources rather than learning source-specific artifacts.
The implementation of these protocols requires careful planning and execution but enables researchers to leverage the substantial benefits of data pooling while minimizing methodological risks. Through rigorous assessment and appropriate mitigation strategies, morphometric researchers can enhance their understanding of shape space and classification while maintaining analytical integrity.
The selection of anatomical templates and the methods used to register study samples to them are critical steps in geometric morphometrics (GM) that directly influence the accuracy and reliability of subsequent shape classification. Traditional single-template approaches often introduce registration bias, especially when morphological variability is high, which can compromise the analysis of out-of-sample individuals and the general application of classification rules. This technical guide synthesizes current methodologies, advocating for multi-template and functional data approaches to mitigate these issues. Framed within a broader thesis on understanding shape space, this document provides researchers and drug development professionals with advanced protocols to enhance the precision of morphometric classification in biomedical and evolutionary research.
In geometric morphometrics, a template is a reference configuration of landmarks to which all other specimens in a study are aligned. The process of registration involves using algorithms to superimpose these specimens onto the template, removing differences due to position, orientation, and scale to isolate pure shape variation. The geometry of shape space is non-linear and complex; it is a subspace of the original coordinate space, accounting for the fact that configurations differing only by rotation, translation, or scaling represent the same shape [74]. The choice of template and registration protocol directly influences the geometry of this shape space and, consequently, the performance of classifiers built upon it.
The central challenge is that classification rules derived from a sample-dependent shape space cannot be applied to new, out-of-sample individuals in a straightforward manner. Sample-dependent processing steps, such as Generalized Procrustes Analysis (GPA), require the entire sample set for alignment [37]. A poor template choice can lead to registration error, where the alignment process inaccurately represents the true anatomical correspondence between specimens. This error introduces noise and bias into the shape variables, ultimately reducing the classification accuracy of models used to distinguish between groups, such as healthy versus diseased states or different species.
Methodological Approaches to Template Selection and Registration
Single-Template versus Multi-Template Approaches
The conventional approach relies on a single template, which can be an image from a single subject, a population-average template, or a standardized atlas. However, this method is highly susceptible to the specific characteristics of the chosen template. If the template is morphologically distant from a target specimen, registration accuracy diminishes, a problem exacerbated in studies with high morphological variability [75] [76].
Multi-template approaches have been developed to address this limitation. By using multiple templates that collectively represent the morphological diversity of the population, registration errors are averaged and compensated for across different registrations. The underlying assumption is that the biases introduced by individual templates will cancel out, leading to a more robust and accurate final estimate of the true shape.
Table 1: Comparison of Single-Template and Multi-Template Approaches
| Feature | Single-Template Approach | Multi-Template Approach |
|---|---|---|
| Core Principle | All specimens are registered to one reference template. | Specimens are registered to multiple templates; results are combined. |
| Handling of Registration Error | Highly susceptible to bias if the template is not representative. | Averages and compensates for registration errors across templates. |
| Robustness to Variability | Low; performance declines with high sample variability. | High; designed to accommodate diverse morphological forms. |
| Computational Cost | Lower. | Higher, as multiple registrations are required. |
| Best Suited For | Studies with very low intra-sample morphological variance. | Studies with high morphological variability (e.g., evolutionary biology, disease progression). |
Key Multi-Template and Registration Methodologies
Several specific methodologies exemplify the advanced application of multi-template and registration techniques:
- MALPACA (Multiple Automated Landmarking through Point Cloud Alignment and Correspondence): This pipeline uses multiple templates for automated landmarking. For each target specimen, landmark estimates are generated by registering it to every template in the set. The final landmark coordinates are determined by taking the median estimate for each coordinate across all templates. This method significantly outperforms single-template methods in landmarking both single- and multi-species samples [75].
- K-Means Template Selection: A critical step in multi-template analysis is the selection of a representative set of templates. When no prior information is available, a K-means-based method can be used. This involves:
- Performing a Generalized Procrustes Analysis (GPA) on sparse point clouds of the study sample.
- Conducting a Principal Component Analysis (PCA) on the Procrustes coordinates.
- Applying K-means clustering on the PC scores to identify morphological clusters.
- Selecting specimens closest to the cluster centroids as templates, ensuring they capture the population's morphological diversity [75].
- Functional Data Geometric Morphometrics (FDGM): This approach converts discrete 2D landmark data into continuous curves, which are represented as linear combinations of basis functions. This allows for the analysis of shape changes as continuous functions, capturing subtle variations and local deformations that may be missed by traditional landmark-based GM. FDGM has been shown to outperform classical GM in classifying species with subtle craniodental shape differences [77].
- Square-Root Velocity Function (SRVF): A diffeomorphic method that transforms shapes such that the shape space becomes a sphere. This allows for easy computation of distances as great circles and has been demonstrated to outperform other shape description methods, including eigenshapes, in classification tasks involving organic and man-made objects [74].
Experimental Evidence and Quantitative Validation
Validation in Neuroimaging and Alzheimer's Disease Diagnostics
A landmark study on Tensor-Based Morphometry (TBM) for Alzheimer's disease (AD) classification provides compelling quantitative evidence for the multi-template approach. Using 772 subjects from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database and 30 templates, researchers compared single-template and multi-template TBM methods [76].
Table 2: Classification Accuracy of Multi-Template vs. Single-Template TBM in Alzheimer's Disease [76]
| Subject Groups | Single-Template TBM Accuracy | Multi-Template TBM Accuracy |
|---|---|---|
| Control vs. Alzheimer's Disease | Lower than multi-template | 86.0% |
| Stable MCI vs. Progressive MCI | Lower than multi-template | 72.1% |
The study found that the improvement offered by multi-template methods was statistically significant. Furthermore, the statistical group-level difference maps produced with multi-template TBM were smoother, formed larger continuous regions, and had higher t-values, indicating greater sensitivity in detecting morphological changes associated with disease [76].
Validation in Biological Morphometrics
The superiority of multi-template methods extends beyond medical neuroimaging. In a study aimed at automated landmarking of mouse and ape skulls, the MALPACA pipeline was rigorously validated against a "gold standard" of manual landmarks [75].
- Result: MALPACA significantly outperformed single-template methods (ALPACA) as measured by Root Mean Square Error (RMSE) when compared to manual landmarks. The multi-template approach also showed higher correlation with gold-standard morphometric variables such as centroid size and Procrustes distances.
- Protocol: The validation involved landmarking 61 mouse skull models and 52 great ape skull models. The performance was quantified by comparing the automated landmark positions to manually annotated "gold standard" landmarks and comparing the resulting morphometric variables [75].
Similarly, the FDGM approach was tested on three shrew species (S. murinus, C. monticola, and C. malayana) using craniodental landmarks [77].
- Result: When combined with machine learning classifiers (Naïve Bayes, SVM, Random Forest, etc.), the FDGM method achieved higher classification accuracy for the three species compared to classical GM. The dorsal view of the skull was identified as the most informative for classification.
- Protocol: The study used landmark data from 89 shrew specimens. Both GM and FDGM methods were applied, followed by PCA and LDA. Machine learning models were trained on the resulting PC scores to compare classification performance [77].
Practical Protocols for Implementation
A Protocol for Multi-Template Analysis with MALPACA
Diagram 1: MALPACA Workflow
- Template Selection:
- If prior knowledge of morphological variation is available, select templates to cover this range.
- If no prior information exists, use the K-means selection method:
- Extract sparse point clouds from all specimen models.
- Perform GPA and PCA on the point clouds.
- Apply K-means clustering to the PC scores.
- Select specimens nearest to the cluster centroids as templates.
- Template Landmarking: Manually annotate the chosen templates with the required landmarks. This is the only manual step in the pipeline.
- Multiple Registrations: For each target specimen, run the automated landmarking algorithm (e.g., ALPACA) independently for each template. This generates multiple sets of landmark estimates for the target.
- Data Fusion: For each landmark coordinate (x, y, z) in the target specimen, calculate the median value from all estimates provided by the different templates. This median becomes the final coordinate value [75].
A Protocol for Two-Level Deformation-Based Morphometry
For longitudinal studies, a two-level Deformation-Based Morphometry (DBM) pipeline offers superior sensitivity for detecting within-subject changes.
Diagram 2: Two-Level DBM Pipeline
- Level 1 (Within-Subject):
- For each subject with multiple time points, create an unbiased subject-specific template using all available images from that subject.
- Calculate the Jacobian determinants (measuring local volume change) from the deformation fields that map each time-point image to this subject-specific template. This captures longitudinal change within the individual with high accuracy.
- Level 2 (To Common Space):
- Create an unbiased average template for the entire study population.
- Co-register all individual Jacobian maps from Level 1 to this common group space.
- Statistical Analysis: Perform voxel-wise statistical modeling on the co-registered Jacobian maps in the common space to compare morphological changes across subjects or groups. This pipeline has been shown to be 4.5 times more sensitive in detecting longitudinal volume changes compared to one-level DBM [78].
Table 3: Key Software and Methodological Tools for Advanced Morphometrics
| Tool/Resource | Function | Application Context |
|---|---|---|
| MALPACA | An open-source pipeline for multiple-template automated landmarking. | Landmarking highly variable biological samples (e.g., across species). |
| SlicerMorph | An open-source extension for 3D Slicer providing tools for GM, including ALPACA and MALPACA. | 3D morphological analysis and visualization in evolutionary biology and biomedicine. |
| Advanced Normalization Tools (ANTs) | A comprehensive toolkit for biomedical image registration, used in DBM/TBM pipelines. | Neuroimaging analysis, including the two-level DBM pipeline for longitudinal MRI studies. |
| Square-Root Velocity Function (SRVF) | A diffeomorphic method that maps shape space to a sphere for efficient computation of shape distances. | High-accuracy classification of outline shapes from various domains (biology, archaeology). |
| Functional Data Geometric Morphometrics (FDGM) | A method that represents landmark data as continuous curves for analyzing subtle shape variations. | Classifying species with minor morphological distinctions or studying complex shape changes. |
| K-Means Template Selection | An unbiased algorithm for selecting a representative set of templates from a population. | Optimal template selection for multi-template analyses when prior morphological knowledge is limited. |
The selection of templates and the methodology of registration are not mere preliminary steps but are foundational to the validity of morphometric classification. Evidence from both evolutionary biology and clinical neuroimaging consistently demonstrates that moving beyond single-template approaches to embrace multi-template registration and functional data representations yields substantial improvements in classification accuracy and analytical sensitivity. By implementing the advanced protocols outlined in this guide—such as MALPACA for landmarking, two-level DBM for longitudinal studies, and SRVF/FDGM for nuanced shape analysis—researchers can more reliably navigate the complexities of shape space. This enhances the utility of morphometrics as a robust tool for critical applications ranging from taxonomic classification to the identification of disease-specific biomarkers.
Ensuring Rigor: Validation Protocols and Comparative Analysis of Techniques
In geometric morphometrics, the accurate classification of biological shapes—from ancient bones to modern clinical specimens—relies on robust statistical validation frameworks. The central challenge lies in developing models that generalize beyond the specific samples used for training, providing reliable predictions for new, unseen data. Within the context of shape space and classification research, validation methodologies ensure that morphological patterns identified through Procrustes analysis and other morphometric techniques represent true biological signals rather than sample-specific idiosyncrasies. As morphometric applications expand into critical areas including paleoanthropological reconstruction, clinical nutritional assessment, and taxonomic identification, improper validation can compromise research validity and practical applications [79] [18] [20].
This technical guide examines structured approaches for training-test splits and cross-validation specifically adapted to morphometric research. These methodologies address two interconnected challenges: (1) obtaining unbiased performance estimates for shape-based classifiers, and (2) selecting optimal model parameters without inflating perceived accuracy through data leakage. By implementing rigorous validation frameworks, researchers can produce classification rules applicable to out-of-sample individuals—a fundamental requirement for both scientific discovery and applied morphological diagnostics [18] [80].
Foundational Concepts: From Simple Splits to Cross-Validation
The Holdout Method: Train-Validation-Test Splits
The simplest validation approach partitions available data into distinct subsets for training, validation, and testing. In this framework, the training set builds the model, the validation set guides hyperparameter tuning and model selection, and the test set provides a final unbiased evaluation on truly unseen data [79] [81].
Implementation Protocol:
- Randomly split the complete dataset into three subsets: training (typically 70%), validation (15%), and test (15%)
- Train all candidate models on the training set
- Evaluate and compare model performance on the validation set
- Select the best-performing model architecture and hyperparameters
- Retrain the selected model on the combined training and validation sets
- Report final performance metrics from the untouched test set [79]
While straightforward, this approach has significant limitations for morphometric studies. The validation estimate can be highly variable depending on the specific data partition, and with smaller sample sizes—common in morphological studies—reducing the training data by 30% may substantially impact model performance by excluding meaningful morphological variation [79] [82].
K-Fold Cross-Validation: Enhanced Reliability for Limited Samples
K-fold cross-validation (CV) addresses holdout method limitations by repeatedly partitioning the data into complementary training and validation subsets. This approach utilizes the available data more efficiently, making it particularly valuable for morphometric studies with limited specimens [83] [84].
Implementation Protocol:
- Randomly divide the dataset into k folds of approximately equal size
- For each fold iteration:
- Designate the current fold as the validation set
- Combine the remaining k-1 folds into a training set
- Train the model on the training set
- Calculate performance metrics on the validation set
- Compute the average performance across all k iterations [84]
For morphometric applications, k=5 or k=10 are common configurations, though the optimal choice depends on dataset size. Each CV iteration produces a different shape space alignment based on the training subset, then applies this alignment to the validation specimens—properly simulating the processing of new, unseen individuals [18].
Table 1: Comparison of Primary Validation Approaches for Morphometric Analysis
| Method | Key Advantages | Key Limitations | Recommended Context |
|---|---|---|---|
| Single Train-Test Split | Simple, computationally efficient | High variance, optimistic bias in reported metrics | Preliminary exploration with large samples |
| Train-Validation-Test Split | No information leakage to test set | Results depend on specific random split | Large datasets (>20,000 specimens) [85] |
| K-Fold Cross-Validation | Reduced variance, efficient data use | Computational intensity, nested alignment required | Small to medium morphometric datasets [79] [84] |
| Stratified K-Fold CV | Maintains class distribution in splits | Increased implementation complexity | Classification with imbalanced morphological groups [84] |
| Nested Cross-Validation | Unbiased performance estimation with hyperparameter tuning | High computational cost | Final model evaluation and protocol development [79] |
Advanced Validation Frameworks for Morphometric Research
Nested Cross-Validation: The Gold Standard for Model Selection and Evaluation
Nested cross-validation (CV) provides the most rigorous framework for both model selection and performance evaluation, making it particularly valuable for morphometric studies requiring definitive validation of shape-based classification systems [79].
Implementation Protocol:
- Outer Loop: Split data into k-folds for model evaluation
- Inner Loop: For each training set from the outer loop:
- Perform additional k-fold splits
- Train models with different hyperparameters on these inner training sets
- Validate on inner test folds to identify optimal parameters
- Retrain with optimal parameters on the complete inner training set
- Evaluate the resulting model on the outer test fold
- Repeat for all outer folds and average performance metrics [79]
In geometric morphometrics, this approach ensures that Procrustes alignment, allometric correction, and other sample-dependent processing steps are repeatedly recomputed using only training data, properly simulating the application of the final classification rule to new specimens [18].
Figure 1: Nested Cross-Validation Workflow for Morphometric Analysis
Special Considerations for Geometric Morphometric Data
Geometric morphometrics introduces unique validation challenges not typically encountered in standard machine learning applications. The requirement for Generalized Procrustes Analysis (GPA) to align specimens into shape space before classification means that validation frameworks must properly account for this sample-dependent processing [20] [80].
Critical Validation Protocol Adjustments:
Alignment Independence: Procrustes alignment must be computed exclusively from training data in each validation fold, with validation specimens projected into the resulting shape space using the training-derived alignment parameters. This prevents information leakage from validation specimens influencing the shape space construction [18].
Allometric Correction: When applying size correction (allometric regression), regression parameters must be derived from training data only and applied to validation specimens. This ensures the correction generalizes to new individuals [18].
Template Registration: For semi-landmark and surface analysis, registration templates must be defined using training specimens. Out-of-sample individuals are then registered to these training-derived templates [18] [20].
Missing Data Imputation: For incomplete morphological specimens, imputation methods must be trained exclusively on training data, with these models applied to missing data in validation specimens [20].
Table 2: Morphometric-Specific Processing in Validation Frameworks
| Processing Step | Standard Approach | Proper Validation Protocol | Rationale |
|---|---|---|---|
| Procrustes Alignment | Align all specimens together | Align training set, rotate validation set to training consensus | Prevents information leakage from test specimens |
| Allometric Regression | Compute regression on full sample | Compute on training, apply to validation | Ensures size correction generalizes to new specimens |
| Semilandmark Sliding | Slide all curves/surfaces together | Define template from training, register validation to it | Maintains biological homology across samples |
| Missing Landmark Estimation | Impute using full sample patterns | Train imputation on training, apply to validation | Prevents artificial inflation of performance metrics |
Experimental Protocols and Implementation Guidelines
Case Study: Validation Framework for Taxonomic Identification
Recent research on leaf-footed bugs (genus Acanthocephala) demonstrates proper validation implementation for taxonomic identification. The study employed geometric morphometrics of pronotum shape to discriminate among 11 species, several of quarantine significance [80].
Experimental Protocol:
Specimen Imaging and Landmarking:
- Acquired high-definition images from USDA identification database
- Digitized 40 homologous landmarks on pronotum using TPSDig2 v2.17
- Verified species identification through taxonomic experts [80]
Validation Framework Design:
- Implemented k-fold cross-validation with k=5
- For each fold: Procrustes alignment computed from training specimens only
- Validation specimens projected into training-derived shape space
- Principal Component Analysis (PCA) on training covariance matrix
- Classification using discriminant analysis [80]
Performance Metrics:
- Classification accuracy averaged across folds
- Mahalanobis distances between species groups
- Procrustes ANOVA for shape significance testing [80]
This validation approach confirmed that pronotum shape provides reliable species discrimination, with significant Mahalanobis distances between most species pairs. The rigorous cross-validation protocol ensured that classification accuracy estimates realistically represented performance on genuinely new specimens [80].
Case Study: Nutritional Status Classification from Body Shape
Research on children's nutritional status classification demonstrates validation considerations for clinical morphometric applications. The study addressed the critical challenge of classifying out-of-sample individuals not included in the original study sample [18].
Methodological Framework:
Data Acquisition:
- Collected body shape images from reference population
- Established standardized photographic protocols
Validation Strategy:
- Nested cross-validation for model selection and evaluation
- Multiple template configurations tested for registration of out-of-sample individuals
- Analysis of template selection impact on classification performance [18]
Key Findings:
- Template configuration significantly influences out-of-sample classification
- Sample characteristics and collinearity among shape variables crucial for performance
- Validation framework enabled updating training samples across different screening campaigns [18]
This approach highlights how proper validation protocols enable the development of morphometric classification systems deployable in practical clinical contexts, such as the SAM Photo Diagnosis App for nutritional assessment [18].
Figure 2: Comprehensive Morphometric Validation Workflow
Table 3: Essential Software and Methodological Tools for Morphometric Validation
| Tool Category | Specific Solutions | Function in Validation | Implementation Considerations |
|---|---|---|---|
| Shape Analysis Software | MorphoJ, geomorph R package | Procrustes alignment, PCA, discriminant analysis | Batch processing for cross-validation folds [86] [80] |
| Landmark Digitization | TPSDig2, Viewbox 4 | Coordinate data acquisition | Consistent landmark protocols across samples [20] [80] |
| Machine Learning Frameworks | scikit-learn, R caret | Cross-validation implementation | Integration with morphometric data structures [83] [85] |
| Statistical Analysis | R, PAST, Python SciPy | Procrustes ANOVA, multivariate statistics | Automation for repeated validation trials [86] |
| Data Management | Pandas, R data tables | Handling training/validation splits | Tracking specimen metadata across folds [82] [85] |
Validation through appropriate training-test splits and cross-validation represents a methodological cornerstone for morphometric classification research. As geometric morphometrics expands into increasingly impactful applications—from taxonomic identification with agricultural and quarantine significance to clinical nutritional assessment—rigorous validation ensures that shape-based classification rules generalize beyond the specific specimens studied. The frameworks outlined in this guide provide structured approaches for obtaining unbiased performance estimates while properly accounting for the unique characteristics of morphometric data, particularly the sample-dependent nature of Procrustes alignment and shape space construction.
By implementing these validation protocols, researchers can develop morphological classifiers with demonstrated reliability for new specimens, advancing both scientific understanding of shape variation and practical applications across biological, anthropological, and clinical domains.
The quantitative analysis of shape is a cornerstone of research across biological, geological, and medical sciences. The selection of an appropriate shape space model and classifier is pivotal, as it directly influences the accuracy, interpretability, and scalability of morphological studies. This whitepaper provides an in-depth comparative analysis of predominant methodologies in morphometrics, from traditional geometric morphometrics to modern deep learning approaches. We synthesize performance data from diverse applications—including paleontology, archaeobotany, and medical diagnostics—to evaluate models based on classification accuracy, computational efficiency, and robustness. Furthermore, we present standardized experimental protocols and a curated toolkit to guide researchers in selecting and implementing optimal analytical frameworks for their specific research questions, thereby advancing the reproducibility and rigor of shape-based classification in scientific research.
Shape is a fundamental property of objects, and its quantification is essential for tasks ranging from fossil identification and clinical diagnosis to evolutionary biology [21] [3]. The field of morphometrics provides the theoretical and practical tools for this quantification, primarily through the construction of shape spaces—mathematical spaces where each point represents a distinct object shape. The choice of a shape space model, coupled with a classification algorithm, defines an analytical pipeline whose performance determines the validity of scientific inferences [26] [9].
Historically, geometric morphometrics (GM) , particularly those based on expert-placed landmarks, has been the dominant framework. While powerful, these methods are often constrained by manual digitization, which introduces observer bias and limits the scope of analyzable morphological features [24]. The burgeoning availability of 3D data and computational power has catalyzed the development of automated and "landmark-free" approaches. These include automated geometric morphometric pipelines like auto3DGM and morphVQ [24], as well as deep learning (DL) models that learn shape features directly from images [87] [88].
This review is framed within a broader thesis that understanding the comparative performance of these evolving methodologies is critical for the advancement of morphometrics research. We move beyond a simple catalog of methods to a rigorous, evidence-based comparison of their performance across different domains, providing researchers with a clear guide for navigating the complex landscape of modern shape analysis.
Theoretical Foundations of Shape Spaces
At its core, a shape space is a manifold where geometrical objects are represented, and distances between points correspond to a quantitative measure of shape dissimilarity [26]. The structure of this space is determined by the chosen shape representation and correspondence model.
Key Shape Space Models
Procrustes Shape Space: This is a foundational model in traditional GM. Shapes are represented by configurations of landmarks (anatomically defined homologous points). Through Generalized Procrustes Analysis (GPA), configurations are superimposed to remove the effects of translation, rotation, and scale. The resulting Procrustes coordinates reside in a non-Euclidean curved space, though tangent space projections are typically used for multivariate statistical analysis [9] [17]. This model explicitly requires a priori biological knowledge to define landmarks.
Form Space: In contrast to the pure shape space of Procrustes analysis, form space retains size information. This aligns with the Huxley–Jolicoeur school of allometry, which studies covariation among morphological features that all contain size information, without a strict separation of size and shape [9]. Analysis in form space often uses Principal Component Analysis (PCA), where the first principal component frequently captures allometric trends.
Functional Map Space: This modern approach represents shape correspondence as a linear map between functions defined on two surfaces. Methods like
morphVQuse descriptor learning to estimate these functional maps between whole 3D meshes, capturing continuous correspondences without predefined landmarks. The shape variation is then quantified through latent shape space differences (LSSDs), providing a comprehensive representation of morphological variation [24].Deep Learning Feature Space: In deep learning, particularly with Convolutional Neural Networks (CNNs), the shape space is implicitly defined by the activations of a network layer. The model learns a hierarchical representation of shape from raw pixels, and the high-dimensional feature vector extracted from a penultimate layer serves as a point in a deep learning feature space. This space is optimized for the specific classification task during training [87] [88].
The Role of Allometry
Allometry—the study of size-related shape changes—is a critical consideration in shaping the space. The Gould–Mosimann school defines allometry as the covariation of shape with size, typically analyzed through the multivariate regression of Procrustes shape coordinates on a size proxy like centroid size [9]. The choice to analyze data in shape space (size removed) versus form space (size retained) will fundamentally alter the resulting ordination and the biological interpretations of allometric patterns.
Methodological Approaches and Classifiers
Shape analysis pipelines can be broadly divided into two paradigms: those that extract hand-crafted shape features and those that learn features directly from data.
Traditional and Automated Morphometric Pipelines
These methods involve an explicit feature extraction step before classification.
- Landmark-based Geometric Morphometrics: This is the traditional workflow involving manual digitization of landmarks, GPA, and then statistical analysis of the Procrustes coordinates [24]. Classification is typically performed using linear discriminant analysis (LDA), logistic regression, or support vector machines (SVM) [17].
- Outline-Based Methods: For structures lacking discrete landmarks, outlines can be analyzed using methods like Elliptical Fourier Analysis (EFA) or eigenshape analysis. These methods transform a closed contour into a set of Fourier coefficients or other shape functions that serve as input for classifiers [87] [3].
- Automated 3DGM Pipelines:
auto3DGMandmorphVQrepresent advances in automating 3D shape analysis.auto3DGMuses farthest point sampling to subsample 3D meshes and then establishes correspondence via a Generalized Dataset Procrustes Framework.morphVQleverages machine learning to compute functional maps between entire surfaces, characterizing shape variation through area-based and conformal latent shape space differences [24].
Deep Learning Approaches
Deep learning models, particularly CNNs and Vision Transformers (ViTs), integrate feature extraction and classification into a single end-to-end learning process.
- Convolutional Neural Networks (CNNs): Models like ResNet and EfficientNet learn hierarchical features directly from images. They have demonstrated superior performance in tasks like archaeobotanical seed classification and skin lesion diagnosis [87] [88].
- Vision Transformers (ViTs): More recently, transformer-based architectures have been applied to visual tasks. They use a self-attention mechanism to model global dependencies in the image. Studies in skin lesion classification have shown that ViTs like Swin-Tiny can achieve state-of-the-art performance and produce more interpretable saliency maps [88].
Classifiers for Morphometric Data
The choice of classifier is often tied to the feature extraction method.
- Linear Discriminant Analysis (LDA): Commonly used with Procrustes coordinates for its simplicity and effectiveness in finding linear combinations that separate groups.
- Support Vector Machines (SVM): Effective for both linear and non-linear classification problems, especially with high-dimensional data.
- Random Forests and XGBoost: Powerful ensemble methods that can capture complex interactions, sometimes used with pre-extracted morphometric features [88].
- Softmax Classifier: The standard final layer for deep learning models, which outputs a probability distribution over the target classes.
Comparative Performance Analysis
A synthesis of recent studies reveals a nuanced performance landscape where no single method is universally superior, but clear trends emerge based on data type and problem context.
Table 1: Comparative Performance of Shape Analysis Models Across Disciplines
| Application Domain | Model / Pipeline | Key Performance Metric | Reported Performance | Key Advantage |
|---|---|---|---|---|
| General 3D Morphometrics [24] | Manual Landmark GM | Genus-level classification accuracy | Comparable to auto3DGM/morphVQ | Theoretical grounding in homology |
auto3DGM |
Genus-level classification accuracy | Comparable to manual GM | Automation; avoids observer bias | |
morphVQ (proposed) |
Genus-level classification accuracy | Comparable to manual GM & auto3DGM | Computational efficiency; captures whole-surface variation | |
| Archaeobotany [87] | Outline Analysis (Momocs) | Classification accuracy | Outperformed by CNN | Standardized morphometric workflow |
| Convolutional Neural Network | Classification accuracy | Superior performance | High accuracy; end-to-end learning | |
| Medical Imaging [88] | CNN (e.g., ResNet) | Accuracy (9-class skin lesions) | High (SOTA) | Strong feature extraction |
| Vision Transformer (Swin-Tiny) | Accuracy (9-class skin lesions) | 78.2% (Best) | Modeling long-range dependencies; more interpretable saliencies | |
| Human Perception [21] | Pixel-based metrics | Correlation with human judgments | Low | Simple baseline |
| State-of-the-art CNN | Correlation with human judgments | Moderate | Learned features | |
| ShapeComp (Multi-descriptor) | Correlation with human judgments | High (r=0.63, p<0.01) | Psychophysically validated; integrates multiple shape aspects |
Key Performance Trends
Deep Learning for Raw Image Classification: In tasks involving direct classification from 2D images, such as identifying seeds or skin lesions, deep learning models consistently outperform traditional morphometric methods. A seminal study on archaeobotanical seeds found that CNNs achieved higher classification accuracy than outline-based GM (using Elliptical Fourier Analysis) for distinguishing wild and domestic subspecies [87]. This performance advantage is attributed to the ability of DL models to learn discriminative features directly from data without being constrained by a pre-defined geometric model.
Traditional GM for Hypothesis-Driven Morphology: When the research question involves testing specific hypotheses about predefined anatomical structures, landmark-based GM remains a powerful and interpretable tool. Its strength lies in its foundation in biological homology, allowing for direct visualization of shape changes in anatomical space [9].
The Rise of Automated 3DGM: For comprehensive analysis of complex 3D structures where manual landmarking is impractical, automated pipelines like
morphVQoffer a compelling balance. They capture more morphological detail than sparse landmark sets and achieve comparable classification accuracy to manual GM while being more computationally efficient and less biased [24].Interpretability and Alignment with Human Perception: While DL models can be "black boxes," explainable AI (XAI) techniques like saliency maps are improving interpretability. Notably, a morphometric analysis of these maps found that correct predictions in transformer models were associated with more concentrated and symmetric saliency maps [88]. Furthermore, models combining multiple hand-crafted shape descriptors (
ShapeComp) have been shown to best predict human visual shape similarity judgments, outperforming both pixel-based metrics and standard CNNs [21].
Table 2: Qualitative Strengths and Weaknesses of Different Approaches
| Model Category | Strengths | Weaknesses |
|---|---|---|
| Landmark-based GM | High biological interpretability; well-established statistical framework; tests explicit hypotheses. | Labor-intensive; observer bias; limited to landmarks, missing other shape data. |
| Automated 3DGM (morphVQ) | Comprehensive surface analysis; reduces bias; computationally efficient. | Correspondence may not reflect biological homology; complex implementation. |
| Deep Learning (CNN/ViT) | High accuracy; end-to-end learning; minimal feature engineering; robust to image noise. | "Black-box" nature; requires large datasets; computationally intensive to train. |
| Multi-Descriptor (ShapeComp) | High correlation with human perception; interpretable features; perceptually uniform spaces. | Limited to 2D contours/silhouettes; may not be optimal for all classification tasks. |
Experimental Protocols for Model Evaluation
To ensure robust and reproducible comparisons, researchers should adhere to standardized experimental protocols. The following workflow outlines key steps for a typical image-based classification study.
Figure 1: Workflow for comparative evaluation of shape classification models.
Data Preprocessing and Partitioning
- Image Preprocessing: Standardize images by resizing to a uniform resolution, applying grayscale normalization, and potentially removing background noise. For 3D meshes, preprocessing may include hole filling and mesh simplification [24] [88].
- Critical Protocol: Out-of-Sample Testing: A fundamental challenge in GM is the evaluation of new individuals not included in the initial study sample. The standard practice of performing Generalized Procrustes Analysis (GPA) on the entire dataset before splitting into training and test sets introduces bias, as the test shapes influence the alignment. A robust protocol involves:
- Performing GPA only on the training set to define the shape space.
- Registering each out-of-sample (test) individual to a template or the mean shape of the training set.
- Projecting the registered test specimen into the training set's shape space for classification [17].
- Data Augmentation: For deep learning models, augment the training data with random rotations, flips, scaling, and brightness adjustments to improve model generalization and prevent overfitting [87] [88].
Model Training and Validation
- Cross-Validation: Use k-fold cross-validation or leave-one-out cross-validation (LOOCV) to assess model stability, especially with smaller datasets common in morphological studies [17].
- Performance Metrics: Report a comprehensive set of metrics beyond simple accuracy. These should include:
- Statistical Testing: Employ appropriate statistical tests (e.g., paired t-tests, McNemar's test) to determine if performance differences between models are statistically significant.
The Scientist's Toolkit: Essential Research Reagents
Implementing the aforementioned protocols requires a suite of software tools and resources. The following table details key solutions for building a shape analysis pipeline.
Table 3: Key Research Reagent Solutions for Shape Analysis
| Tool / Resource | Type | Primary Function | Reference / Availability |
|---|---|---|---|
| Momocs | R Package | Outline and landmark-based geometric morphometrics analysis. | [87] |
| morphVQ | Software Pipeline | Automated 3D morphological phenotyping using functional maps. | Code: github.com/oothomas/morphVQ [24] |
| ShapeComp | Model/Code | Quantifies 2D shape similarity from silhouettes using multiple descriptors. | [21] |
| ISIC Archive | Data Repository | Public repository of dermoscopic images for benchmarking medical AI. | [88] |
| Global & Local Statistical Shape Models | Data/Code | Pre-trained 3D statistical models (e.g., for faces) for model fitting. | Published with [26] |
| Grad-CAM++ / LayerCAM | Software Library | Generates saliency maps for explaining predictions of CNN models. | [88] |
The evaluation of shape space models and classifiers is not a search for a single "best" method, but rather a process of matching the analytical tool to the specific research question, data type, and required level of interpretability. Traditional geometric morphometrics remains indispensable for hypothesis-driven studies of homologous structures. In contrast, deep learning models currently offer superior performance for image-based classification tasks where the goal is accurate prediction rather than explicit morphological description [87]. For comprehensive 3D analysis, automated pipelines like morphVQ provide a powerful and efficient middle ground, capturing extensive morphological detail while reducing manual bias [24].
Future progress in the field will depend on increased emphasis on reproducibility and open science. A recent review of machine learning in paleontology found that only 34.3% of studies were fully reproducible, with fewer than 60% sharing their data or code [29]. Adopting the standardized protocols and rigorous benchmarking outlined in this whitepaper will be crucial for advancing our understanding of shape and its implications across the scientific spectrum.
The accurate evaluation of virtual screening (VS) performance is a critical component in both computational drug discovery and morphometrics research. In drug discovery, VS methods sift through vast chemical libraries to identify promising compounds, and their success hinges on rigorous validation protocols [89]. Similarly, in morphometrics, which involves the quantitative analysis of shape and form, the ability to classify new, unseen specimens based on a reference sample is fundamental for applications ranging from evolutionary biology to nutritional assessment [90] [17]. Both fields grapple with a common challenge: ensuring that models and classifiers built on a training set perform reliably on new, out-of-sample data. This guide provides an in-depth technical examination of the core validation paradigms—retrospective and prospective—framed within the unifying context of shape space and classification. It aims to equip researchers with the methodologies and metrics needed to critically assess and advance the state of their screening and classification tools.
The concept of shape space, as defined by geometric morphometrics, provides a powerful framework for this discussion. In morphometrics, objects are represented by configurations of landmarks, and their shapes are compared in a specialized space after procedures like Generalized Procrustes Analysis (GPA) remove differences due to location, scale, and orientation [20]. The challenge of classification emerges when one seeks to assign a new, out-of-sample individual to a group (e.g., a nutritional status or a species) based on its position in this shape space. This process is not straightforward, as the new individual's raw coordinates must first be registered into the shape space of the training sample before the classification rule can be applied [17]. This mirrors the fundamental challenge in virtual screening: a model trained on known active and inactive molecules must be able to accurately rank never-before-seen compounds. Thus, whether classifying a child's nutritional status from arm shape or identifying a drug candidate by its complementarity to a protein target, the principles of robust validation are universally critical.
Theoretical Foundations: Validation in Context
Defining the Validation Paradigms
Retrospective validation is the process of validating a system or process after it has already been implemented and is in operational use, using accumulated historical data [91] [92]. In the context of virtual screening and classifier development, it involves using a benchmark dataset with known outcomes (e.g., active and decoy molecules, or pre-classified skeletal remains) to assess how well a model would have performed. Its primary purpose is to provide an initial, cost-effective estimate of model performance and to select the best model from a set of candidates before committing to costly prospective testing [93] [89]. However, it can be susceptible to biases in benchmark datasets and may not always generalize to real-world scenarios.
Prospective validation, in contrast, is the ultimate test of a model's utility. It involves testing the model's predictions on genuinely new data in a real-world setting. In drug discovery, this means running a virtual screen on a novel compound library and then experimentally validating the top-ranked hits in the laboratory to confirm binding and activity [89]. In morphometrics, it entails using a previously developed classifier to assess the nutritional status or taxonomy of a newly encountered specimen from a different population or archaeological site [17]. Prospective validation is the definitive proof of a model's predictive power and operational effectiveness.
The Language of Shape and Classification
To understand validation in morphometrics, a grasp of core geometric morphometric concepts is essential:
- Landmarks and Semilandmarks: Biologically homologous points (landmarks) and points used to capture the geometry of curves and surfaces (semilandmarks) that digitize an object's shape [20].
- Generalized Procrustes Analysis (GPA): An iterative least-squares optimization process that superimposes shape configurations by removing differences due to location, scale, and orientation. This aligns all specimens into a common coordinate system, known as Kendall's shape space [20].
- Shape Space and Tangent Space: Kendall's shape space is a non-Euclidean space where each point represents a unique shape. For statistical convenience, shapes are often projected into a linear Euclidean tangent space centered at a reference shape (like the mean shape) [20].
- Out-of-Sample Classification: The process of classifying a new individual that was not part of the original (training) sample used to build the classifier. This requires projecting the new individual's raw coordinates into the pre-existing shape space of the training set, a process that is not always straightforward and requires careful methodological consideration [17].
Retrospective Validation: Methods and Metrics
Retrospective validation relies on carefully constructed benchmark datasets and a suite of quantitative metrics to evaluate performance.
Key Experimental Protocols
A standard retrospective validation protocol involves several key steps, applicable to both virtual screening and morphometric classification:
Dataset Curation: The foundation of any retrospective study is a high-quality benchmark.
- For virtual screening, this typically involves a protein target with a set of known active molecules and a set of decoys—molecules that are physicochemically similar but presumed to be inactive [93] [89]. Common benchmarks include DUD-E and CASF.
- For morphometric classification, a reference sample with known group affiliations (e.g., species, nutritional status) is required. For example, a study might use a sample of children with known nutritional status (Severe Acute Malnutrition vs. Optimal Nutritional Condition) based on traditional anthropometric measures [17].
Model Training and Pose Generation: The virtual screening software or classification algorithm is used to generate predictions (binding poses and scores, or group classifications) for every molecule or specimen in the benchmark set.
Performance Calculation: Predictions are compared against the ground truth to calculate validation metrics.
A critical consideration, especially for machine learning models, is the strict separation of data to avoid data leakage. This means the benchmark must be structurally dissimilar to any data used during the model's training phase. The BayesBind benchmark, for instance, was created specifically for this purpose, composed of protein targets distinct from those in its corresponding training set [93].
Core Performance Metrics
The following table summarizes the key metrics used in retrospective validation.
Table 1: Key Metrics for Retrospective Validation
| Metric | Formula/Description | Interpretation | Use Case |
|---|---|---|---|
| Enrichment Factor (EFχ) | EFχ = (Number of actives in top χ% / Total actives) / χ% |
Measures how much better a model is at identifying actives early in the ranked list compared to random selection. An EF of 1 indicates random performance. | Virtual Screening [93] [89] |
| Bayes Enrichment Factor (EFBχ) | EFBχ = (Fraction of actives above score threshold) / (Fraction of random molecules above threshold) |
An improved metric that uses random compounds instead of decoys, avoiding the assumption that decoys are truly inactive. It does not have a hard maximum value [93]. | Virtual Screening [93] |
| Maximum Bayes EF (EFmaxB) | The maximum value of EFBχ achieved over the measurable range. |
Provides a single, optimistic estimate of a model's potential performance in a real-life, very large library screen [93]. | Virtual Screening [93] |
| Area Under the Curve (AUC) | Area under the Receiver Operating Characteristic (ROC) curve. | Measures the overall ability of the model to discriminate between active and inactive compounds across all possible classification thresholds. A value of 0.5 is random, 1.0 is perfect. | Virtual Screening & Morphometrics [89] |
| Classification Accuracy | (True Positives + True Negatives) / Total Population |
The overall proportion of correct classifications. | Morphometrics [17] |
To illustrate how these metrics are used in practice, the table below shows a comparative analysis of different virtual screening methods on the DUD-E benchmark.
Table 2: Example Virtual Screening Performance on DUD-E Benchmark (Median Values) Data adapted from Sunseri & Koes (2024) [93]
| Model | EF₁% | EFB₁% | EF₀.₁% | EFB₀.₁% | EFmaxB |
|---|---|---|---|---|---|
| Vina | 7.0 | 7.7 | 11 | 12 | 32 |
| Vinardo | 11 | 12 | 20 | 20 | 48 |
| Dense (Pose) | 21 | 23 | 42 | 77 | 160 |
A Note on Morphometric Classifier Validation
In morphometrics, a common approach is to use leave-one-out cross-validation: the classifier is built on all but one specimen in the reference sample, and the left-out specimen is classified. This process is repeated for every specimen [17]. This provides a robust retrospective estimate of the classifier's accuracy. However, as previously noted, applying this classifier to a truly new, out-of-sample individual requires a method to project that individual's raw landmark coordinates into the pre-existing shape space of the training sample, which can be done by registering the new configuration to a template or the mean shape of the training set [17].
Prospective Validation: The Ultimate Test
Prospective validation moves beyond historical benchmarks to test a model in a real-world, operational environment.
Protocol for Prospective Validation
A generalized protocol for a prospective virtual screening campaign or a morphometric field study is as follows:
- Model Finalization: Lock the computational model or classifier based on retrospective results.
- Novel Data Collection:
- Prediction and Selection: Use the model to rank the novel library and select a manageable number of top-ranking hits or classifications.
- Experimental Verification:
- For VS: Acquire the physical compounds and test them in biochemical or cellular assays (e.g., binding affinity, functional activity) to confirm the predicted bioactivity [89].
- For Morphometrics: For a classifier, the "verification" may involve comparing the classification to other established methods or waiting for subsequent clinical or morphological confirmation.
- Analysis of Success: The key metric is the hit rate, calculated as
(Number of confirmed active compounds / Total number tested) * 100%. A high hit rate validates the entire screening pipeline.
Case Study: RosettaVS and KLHDC2
A compelling example of a successful prospective validation is the discovery of ligands for the ubiquitin ligase KLHDC2 using the RosettaVS method [89].
- Method: The researchers developed an AI-accelerated virtual screening platform (OpenVS) that used active learning to efficiently screen a multi-billion compound library.
- Prospective Test: They screened this library against KLHDC2 and selected a handful of top-ranked compounds for experimental testing.
- Result: They discovered a compound with single-digit micromolar binding affinity. Furthermore, they solved the X-ray crystallographic structure of the complex, which confirmed that the predicted binding pose was remarkably accurate [89].
- Significance: This end-to-end process, from computational prediction to experimental validation and structural confirmation, provides the highest level of evidence for a virtual screening method's efficacy. The entire screening process was completed in less than seven days, demonstrating the power of modern, high-performance computing approaches [89].
This section details key computational and material resources essential for conducting validation studies in virtual screening and morphometrics.
Table 3: Essential Research Reagents and Resources
| Category | Item | Function & Description |
|---|---|---|
| Virtual Screening Software | RosettaVS [89], AutoDock Vina [89] | Physics-based docking programs used to predict how a small molecule (ligand) binds to a protein target and to score the strength of that interaction. |
| Benchmark Datasets | DUD-E [93] [89], CASF [93] [89], LIT-PCBA [93] | Curated public datasets containing protein targets, known active compounds, and decoy molecules. Used for retrospective validation and benchmarking of new VS methods. |
| Morphometrics Software | R (with geomorph, Morpho) [17], Viewbox [20], 3D Slicer [90] | Software environments for performing geometric morphometric analyses, including landmark digitization, Procrustes alignment, and statistical shape analysis. |
| Validation & Analysis Platforms | OpenVS [89], BayesBind Benchmark [93] | Specialized platforms for running large-scale virtual screens and for fairly evaluating machine learning models on structurally dissimilar test targets. |
| Experimental Assays | Binding Affinity Assays (e.g., SPR, ITC) [89], X-ray Crystallography [89] | Biochemical and biophysical methods used for the experimental confirmation of computational predictions during prospective validation. |
| Data Collection Hardware | Structured-Light 3D Scanner (e.g., Artec Eva) [20], CT Scanners [90] | Hardware used to capture high-resolution 3D shape data of biological specimens (e.g., bones, arms) for morphometric analysis. |
Integrated Workflow: From Retrospective Benchmarking to Prospective Application
The following diagram synthesizes the concepts and methodologies discussed in this guide into a cohesive, end-to-end workflow for model development and validation. It highlights the critical, iterative feedback loop between retrospective analysis and prospective application, which is fundamental to advancing the state of the art in both virtual screening and morphometrics.
Robust validation is the cornerstone of reliable scientific discovery in computational fields. As this guide has detailed, retrospective validation provides an essential, efficient first pass for benchmarking and refining models using historical data. However, it is the rigorous application of prospective validation—testing models against genuinely new data and experimentally verifying the predictions—that separates promising computational tools from those that deliver real-world impact. The unifying framework of shape space and classification elegantly ties together the challenges faced in disparate fields, from identifying a new drug candidate to assessing a child's nutritional status. By adhering to the detailed protocols, metrics, and integrated workflow outlined herein, researchers can ensure their virtual screening and classification approaches are not only statistically sound but also truly predictive and actionable.
In morphometrics research, the quest to quantitatively understand shape space and classification is fundamental. This field, which bridges biology, archaeology, and medicine, relies on computational tools to extract meaningful patterns from complex morphological data. The choice of computational tool directly impacts the reliability, efficiency, and scope of scientific insights. As noted by Nature Biomedical Engineering, thorough benchmarking is a sign of a healthy research ecosystem and is crucial for clarifying a study's potential impact [94]. This guide provides a structured framework for evaluating computational tools used in morphometrics based on the core metrics of speed, accuracy, and user-accessibility, ensuring that research in shape analysis is both robust and reproducible.
Core Metrics for Benchmarking Computational Tools
Evaluating computational tools requires a balanced assessment of three interdependent performance categories.
Accuracy measures the correctness and relevance of a tool's outputs. In morphometrics, this extends beyond simple metrics to include performance on specific tasks like segmentation, classification, and feature extraction. Key metrics include:
- Tool Calling Accuracy: The system's ability to invoke the correct functions or data sources; top tools achieve ≥90% accuracy [95].
- Context Retention: Particularly for AI-powered platforms, the ability to maintain understanding across multi-step queries, with benchmarks also set at ≥90% [95].
- Statistical Performance: Standard metrics like Area Under the Curve (AUC), Dice scores for segmentation, and Hausdorff distances provide quantitative measures of performance, as seen in biomedical tool evaluations [96].
Speed encompasses both computational efficiency and workflow velocity.
- Response Time: The time from query submission to result display. Industry benchmarks target under 1.5 to 2.5 seconds for a seamless user experience [95].
- Update Frequency: How quickly new or modified information becomes available for analysis. Real-time or near-real-time indexing is essential in fast-moving research environments [95].
- Runtime & Hardware Requirements: Computational papers should explicitly report runtimes and computing hardware needs to facilitate fair comparisons [94].
User-Accessibility determines how readily researchers can adopt and utilize a tool effectively.
- Interface Intuitiveness: Measures how quickly new users become productive, with clean navigation and clear visual hierarchies being critical [95].
- Implementation Complexity: The technical expertise required for setup and daily operation, ranging from code-intensive platforms to those with scriptless interfaces [97].
- Reporting Quality: The availability of customizable reports that visualize results and track adoption metrics across user groups [95].
Table 1: Core Metric Benchmarks for Computational Tools
| Metric Category | Specific Metrics | Benchmark Standards | Research Context Examples |
|---|---|---|---|
| Accuracy | Tool Calling Accuracy | ≥90% [95] | Function selection in analysis pipelines |
| Context Retention | ≥90% [95] | Multi-step morphological analyses | |
| Statistical Performance (AUC, Dice Score) | Varies by task (e.g., AUC >0.8) [96] | Seed classification, vessel segmentation | |
| Speed | Response Time | <1.5-2.5 seconds [95] | Querying large morphological databases |
| Update Frequency | Real-time to near-real-time [95] | Incorporating new data into analyses | |
| Runtime/Hardware Needs | Explicit reporting required [94] | Processing large image datasets | |
| User-Accessibility | Interface Intuitiveness | Qualitative usability assessment [95] | Software adoption across research teams |
| Implementation Complexity | Scriptless to code-intensive options [97] | Deployment in diverse research environments | |
| Reporting Quality | Customizable, actionable insights [95] | Publication-ready figure generation |
Benchmarking Methodologies and Experimental Design
Structured Benchmarking Workflow
A rigorous benchmarking process follows a systematic workflow to produce comparable, actionable results:
- Define Objectives and Metrics: Identify the primary research questions and select the most relevant metrics. If reducing analysis time is the goal, prioritize speed and interface usability over exhaustive feature comparisons [95].
- Select and Curate Datasets: Use real-world datasets that reflect actual research scenarios. Data curation should include standardization, removal of inorganic compounds, neutralization of salts, and handling of duplicates [98].
- Establish Baselines: Compare new tools against established benchmarks and gold-standard methods relevant to morphometrics research [94].
- Execute in Controlled Environment: Run tests in consistent hardware/software environments to ensure fair comparisons, documenting all configuration details.
- Analyze and Interpret Results: Evaluate outcomes against predefined objectives, using statistical methods to determine significance.
Case Study: Archaeobotanical Seed Classification
A landmark study directly compared Geometric Morphometric Methods (GMM) against Convolutional Neural Networks (CNNs) for classifying archaeobotanical seeds, providing a template for rigorous benchmarking in morphometrics [87].
Experimental Protocol:
- Dataset: Over 15,000 seed photographs of wild vs. domestic varieties [87].
- Methods Compared: Traditional GMM using the Momocs R package versus CNN implemented using R (reticulate package) and Python [87].
- Performance Assessment: Classification accuracy across different sample sizes to determine minimal data requirements [87].
- Results: CNNs outperformed GMM in classification accuracy, demonstrating the value of deep learning approaches for complex shape classification tasks [87].
Case Study: Neuropathology Morphometry
Another exemplary benchmarking approach appears in neuropathology, where researchers developed a machine learning-based algorithm (ArtSeg) for quantifying brain arteriolosclerosis [96].
Experimental Protocol:
- Task: Automated morphometric analysis of arteriolosclerotic vessels on whole slide images.
- Validation: Internal hold-out testing, 3-fold cross-validation, and external validation on datasets from multiple brain banks.
- Performance Metrics: AUC-ROC values (0.77-0.92), Dice scores (0.56-0.74), and Hausdorff distances (2.15-7.80) across different validation sets [96].
- Outcome: Successfully derived sclerotic indices and vessel wall measurements comparable to expert assessment [96].
Diagram 1: Benchmarking methodology workflow for tool evaluation.
Comparative Analysis of Computational Approaches
Traditional vs. Machine Learning Methods in Morphometrics
The transition from traditional morphometric approaches to machine learning-based methods represents a significant shift in how researchers analyze shape data.
Traditional Methods like Geometric Morphometric Methods (GMM) have established benchmarks for shape analysis through:
- Landmark-Based Analysis: Precise anatomical point correspondence across specimens.
- Outline Analyses: Using elliptical Fourier transforms to capture shape contours.
- Proven Reliability: Decades of validation across biological disciplines.
Machine Learning Approaches offer alternative paradigms:
- Convolutional Neural Networks (CNNs): Automatically learn relevant features from image data without predefined landmarks.
- Random Forest Algorithms: Effectively handle nonlinear relationships in morphological data, with one study showing R² = 0.84 for predicting morphological traits in plants [99].
- Deep Learning Frameworks: Revolutionize bioimage analysis by integrating segmentation, tracking, and feature extraction [100].
Table 2: Method Comparison in Morphometrics Research
| Method Type | Specific Tools/Approaches | Accuracy Performance | Speed Considerations | Accessibility Requirements |
|---|---|---|---|---|
| Traditional Morphometrics | Geometric Morphometrics (GMM) | Lower than CNN in seed classification [87] | Established, optimized workflows | Requires expertise in shape theory |
| Machine Learning | Convolutional Neural Networks (CNN) | Superior to GMM for classification [87] | Training computationally intensive; fast inference | Coding proficiency often needed |
| Machine Learning | Random Forest (RF) | R² = 0.84 for trait prediction [99] | Efficient for medium-sized datasets | Interpretable, less complex than deep learning |
| Machine Learning | Multi-layer Perceptron (MLP) | R² = 0.80 for trait prediction [99] | Architecture-dependent performance | Requires tuning of hyperparameters |
| Integrated Frameworks | ML + Optimization Algorithms (e.g., RF-NSGA-II) | Enables multi-objective optimization [99] | Additional computational overhead | Combines modeling and decision support |
The Researcher's Toolkit: Essential Software Solutions
Morphometrics research utilizes diverse computational tools, each with distinct strengths and limitations:
Table 3: Computational Tools for Morphometrics Research
| Tool Name | Primary Use Case | Accuracy Features | Speed Performance | Accessibility Level |
|---|---|---|---|---|
| R/Python Ecosystems | Flexible morphometric analyses | High with proper implementation [87] [99] | Varies with implementation | Steep learning curve |
| Apache JMeter | Performance/load testing | Detailed performance metrics [97] [101] | Scalable to heavy loads | GUI and scripting options |
| Gatling | High-performance load testing | Real-time detailed reports [97] [101] | Highly efficient, low resource use | Code-centric (Scala) |
| k6 | Cloud-native testing | JavaScript scripting for customization [101] | Optimized for CI/CD pipelines | Developer-friendly |
| LoadRunner | Enterprise-level testing | Advanced monitoring capabilities [97] | Handles complex, high-volume tests | Commercial, high cost |
Practical Applications in Morphometrics Research
Integrated Framework for Predictive Modeling and Optimization
Advanced morphometrics research increasingly combines machine learning with optimization algorithms to extract maximum insight from shape data. A study on Roselle (Hibiscus Sabdariffa L.) demonstrates this powerful integration:
Experimental Protocol:
- Data Collection: Field experiments measuring morphological traits (branch number, growth period, boll number, seed number) across ten genotypes and five planting dates [99].
- Model Development: Random Forest (RF) and Multi-layer Perceptron (MLP) models trained to predict morphological traits based on genotype and planting date [99].
- Performance Assessment: RF demonstrated superior predictive capability (R² = 0.84) compared to MLP (R² = 0.80) [99].
- Optimization Phase: The trained RF model integrated with Non-dominated Sorting Genetic Algorithm II (NSGA-II) to identify optimal genotype-planting date combinations for maximizing multiple morphological traits simultaneously [99].
- Validation: The Qaleganj genotype planted on May 5 achieved optimal values: 26 branches/plant, 176-day growth period, 116 bolls/plant, and 1517 seeds/plant [99].
This integrated approach highlights how computational tools can advance from simple prediction to prescriptive optimization in morphometrics research.
Diagram 2: ML with optimization for predictive morphometrics.
Essential Research Reagents and Computational Materials
Successful implementation of morphometric analyses requires both wet-lab and computational resources:
Table 4: Essential Research Reagents and Computational Materials
| Item Category | Specific Examples | Function in Research |
|---|---|---|
| Biological Specimens | Roselle genotypes (Qaleganj, HA, HS-24) [99] | Provide morphological variation for analysis |
| Archaeobotanical seed collections [87] | Enable classification algorithm development | |
| Human brain tissue samples [96] | Facilitate neuropathology algorithm validation | |
| Computational Tools | R Statistical Environment with Momocs package [87] | Traditional geometric morphometrics analysis |
| Python with TensorFlow/PyTorch [87] | Deep learning implementation for shape analysis | |
| Image processing libraries (OpenCV, scikit-image) | Preprocessing and feature extraction from images | |
| Validation Frameworks | Cross-validation protocols (k-fold, hold-out) [96] [99] | Ensure model generalizability and prevent overfitting |
| External validation datasets [96] [98] | Test performance on independent data | |
| Applicability domain assessment methods [98] | Determine appropriate scope of model application |
Benchmarking computational tools for speed, accuracy, and user-accessibility is not merely an academic exercise but a fundamental requirement for advancing morphometrics research. As the field progresses toward increasingly complex analyses of shape space and classification, researchers must employ structured benchmarking approaches that directly compare traditional and machine-learning methods using real-world datasets and standardized metrics. The integration of predictive modeling with optimization algorithms represents the cutting edge of computational morphometrics, enabling both understanding of shape variation and identification of optimal outcomes. By adopting rigorous benchmarking practices detailed in this guide, researchers can ensure their computational methodologies are as robust and reproducible as the scientific conclusions they enable.
Reproducibility is a cornerstone of scientific progress, enabling the validation and building upon of previous research findings. In biomedical research, including morphometrics, the reproducibility of experimental findings is essential for them to be broadly accepted as credible by the scientific community [102]. However, for knowledge to be effectively shared and verified, research must be reported with exceptional transparency and rigor. This is particularly crucial in morphometric research, where the analysis of biological shape and form employs sophisticated methodologies that must be precisely documented to enable independent verification. The challenge of reproducibility has prompted major scientific organizations, including the National Academies of Sciences, Engineering, and Medicine, to convene experts for developing better guidelines for transparent reporting [102]. This guide outlines best practices for reporting morphometrics research, with a specific focus on studies investigating shape space and classification, to ensure that findings are both robust and reproducible.
Fundamental Concepts: Shape, Size, and Allometry
Defining Allometry in Geometric Morphometrics
Allometry, a central concept in morphometrics, refers to the size-related changes in morphological traits and remains an essential concept for studying evolution and development [9]. In geometric morphometrics, two primary schools of thought guide allometric studies:
- The Gould-Mosimann School: This perspective defines allometry as the covariation of shape with size. It is implemented statistically through the multivariate regression of shape variables on a measure of size, such as centroid size [9].
- The Huxley-Jolicoeur School: This framework views allometry as the covariation among morphological features that all contain size information. Here, allometric trajectories are characterized by the first principal component in a principal component analysis [9].
Understanding these distinctions is critical for selecting appropriate analytical methods and accurately interpreting results in shape classification studies.
Levels of Allometric Variation
Allometric analyses can be applied at different biological levels, each with distinct implications for research design and interpretation [9]:
- Ontogenetic Allometry: Concerns shape changes associated with growth during individual development.
- Static Allometry: Examines shape covariation with size within a single population at the same developmental stage (typically adults).
- Evolutionary Allometry: Addresses morphological changes associated with size variation across evolutionary lineages.
Each level requires specific sampling strategies and analytical approaches. Confounding these levels by using datasets with multiple sources of size variation can lead to problematic interpretations unless appropriate statistical controls are implemented.
Standards for Transparent Reporting
Guidelines for Reporting Experimental Design
Transparent reporting begins with a comprehensive description of the experimental design, which allows reviewers and other researchers to assess potential biases and the generalizability of findings.
Essential elements to report include:
- Sample Origin and Selection Criteria: Precise documentation of specimen sources, collection methods, and any inclusion/exclusion criteria applied.
- Sample Size Justification: Explanation of how sample sizes were determined, including power analyses where applicable.
- Group Definitions: Clear operational definitions for any biological groups, populations, or taxa included in the study.
- Landmark Selection Protocol: Detailed rationale for landmark choices, including type (anatomical, mathematical, or semi-landmarks) and precise anatomical locations.
- Control Specimens: Identification of any control groups or reference samples used for comparison.
Methodological Documentation for Reproducibility
The methods section must provide sufficient detail to enable exact replication of the analytical procedures. Key components include:
Data Collection Protocols:
- Imaging Specifications: Complete description of imaging equipment, settings, and standardization procedures.
- Landmark Digitization Process: Software used, number of repeated trials, and procedures for minimizing measurement error.
- Data Collection Environment: Conditions under which data were collected to identify potential environmental influences.
Analytical Procedures:
- Software and Versions: Specific software packages, versions, and custom scripts used in analyses.
- Statistical Methods: Complete description of statistical approaches, including any data transformations, algorithms, and significance testing methods.
- Shape Space Definition: Explicit definition of the shape space used (Kendall's shape space, tangent space, etc.) and justification for its selection [103].
- Validation Procedures: Methods used to validate analytical outcomes, such as cross-validation or resampling techniques.
Data Presentation and Visualization
Effective Presentation of Quantitative Data
Proper presentation of quantitative data is fundamental to clear scientific communication. Data should be organized according to their type (categorical or numerical) and presented using appropriate tabular or graphical formats [104].
Table 1: Recommended Data Presentation Formats by Variable Type
| Variable Type | Definition | Recommended Tables | Recommended Charts |
|---|---|---|---|
| Categorical | Characteristics measured by category [104] | Frequency distribution table with absolute/relative frequencies [104] | Bar chart, Pie chart [104] |
| Numerical Discrete | Observations that take certain numerical values [104] | Frequency table with cumulative frequencies [104] | Histogram, Frequency polygon [105] |
| Numerical Continuous | Measurements on a continuous scale [104] | Grouped frequency table with class intervals [105] | Histogram, Frequency curve [106] |
For continuous variables, transformation into categories using class intervals is often necessary. The process should follow these guidelines [105]:
- Calculate the range (highest value - lowest value)
- Divide the range into 5-16 equal intervals
- Define intervals that are mutually exclusive and collectively exhaustive
- Clearly document the boundaries and midpoints of each interval
Table 2: Example Frequency Distribution for a Continuous Morphometric Variable (Centroid Size)
| Class Interval | Absolute Frequency | Relative Frequency (%) | Cumulative Frequency (%) |
|---|---|---|---|
| 10.0 - 12.0 | 5 | 8.3 | 8.3 |
| 12.1 - 14.1 | 12 | 20.0 | 28.3 |
| 14.2 - 16.2 | 18 | 30.0 | 58.3 |
| 16.3 - 18.3 | 15 | 25.0 | 83.3 |
| 18.4 - 20.4 | 10 | 16.7 | 100.0 |
| Total | 60 | 100.0 |
Visualization of Analytical Workflows
Clear visualization of research methodologies helps readers understand complex analytical processes. The following diagram illustrates a standard workflow for a morphometric study investigating allometry and shape classification:
Morphometric Analysis Workflow
For studies involving allometric trajectories, visualizing the relationship between size and shape is particularly important:
Conceptual Approaches to Allometry Analysis
Essential Research Reagents and Materials
Reproducible morphometric research requires precise documentation of all materials and analytical tools. The following table outlines key resources for a comprehensive morphometric study:
Table 3: Research Reagent Solutions for Morphometrics
| Category | Specific Item/Software | Function/Purpose |
|---|---|---|
| Imaging Equipment | Micro-CT Scanner, Digital SLR Camera, Flatbed Scanner | High-resolution specimen imaging for 2D/3D landmark digitization |
| Landmark Digitization | tpsDig2, Viewbox, MorphoJ | Precise landmark coordinate capture and management |
| Shape Analysis Software | MorphoJ, R (geomorph package), PAST | Generalized Procrustes Analysis, shape space construction, and statistical analysis of shape variation |
| Statistical Packages | R, SPSS, PAST | Multivariate statistical analysis, hypothesis testing, and visualization |
| Data Repository | MorphoSource, Dryad Digital Repository | Archiving of raw landmark data and specimen images for verification and reuse |
Experimental Protocols for Key Morphometric Analyses
Protocol 1: Generalized Procrustes Analysis (GPA)
Generalized Procrustes Analysis is the foundational procedure for superimposing landmark configurations prior to shape analysis.
Procedure:
- Landmark Input: Import raw landmark coordinates from all specimens.
- Centering: Translate all configurations to a common origin (usually the centroid).
- Scaling: Normalize each configuration to unit centroid size.
- Rotation: Optimally rotate configurations to minimize the sum of squared distances between corresponding landmarks.
- Procrustes Coordinates: Output the aligned Procrustes coordinates for subsequent analysis.
- Residual Inspection: Examine Procrustes residuals to identify potential outliers or digitization errors.
Documentation Requirements:
- Software and version used for GPA
- Algorithm specifications (e.g., Gower 1975 algorithm)
- Number of iterations required for convergence
- Final Procrustes distance statistics
Protocol 2: Allometry Analysis via Multivariate Regression
This protocol assesses the relationship between shape and size using the Gould-Mosimann framework.
Procedure:
- Size Variable Calculation: Compute centroid size for each specimen.
- Shape Variable Preparation: Extract Procrustes coordinates or principal components from GPA.
- Regression Model: Perform multivariate regression of shape variables on centroid size (log-transformed if necessary).
- Significance Testing: Assess statistical significance using appropriate permutation tests (e.g., 10,000 permutations).
- Visualization: Create visualizations of shape changes associated with size variation (vector displacement diagrams).
- Goodness-of-Fit: Report proportion of shape variance explained by size (R² value).
Documentation Requirements:
- Complete regression statistics
- Permutation test parameters and results
- Visualization parameters and magnification factors for shape changes
Protocol 3: Shape Space Comparison and Classification
This protocol enables the comparison of shapes and classification of unknown specimens into predefined groups.
Procedure:
- Shape Space Definition: Establish the appropriate shape space (Kendall's shape space, tangent space approximation) [103].
- Distance Calculation: Compute Procrustes distances between all specimen pairs.
- Group Discrimination: Perform discriminant analysis or MANOVA to test for significant shape differences between groups.
- Classification Model: Develop classification models (e.g., linear discriminant analysis, random forests) using shape variables.
- Cross-Validation: Implement k-fold cross-validation to assess classification accuracy.
- Visualization: Create scatterplots of principal components or canonical variates to visualize group separation.
Documentation Requirements:
- Shape space model and justification
- Distance matrix characteristics
- Classification model parameters and performance metrics
- Cross-validation protocol and results
Adopting comprehensive reporting standards is essential for advancing morphometric research and ensuring its credibility. By meticulously documenting experimental designs, analytical procedures, and results using the frameworks outlined in this guide, researchers can significantly enhance the reproducibility and robustness of their findings. The specialized nature of shape analysis requires particular attention to methodological transparency, especially in defining shape spaces and allometric relationships. As morphometric techniques continue to evolve and find new applications in evolutionary biology, biomedical research, and drug development, consistent implementation of these reporting practices will facilitate more effective scientific communication and more reliable building of knowledge across the research community.
Conclusion
The integration of shape space theory with robust classification methods provides a powerful framework for quantifying and interpreting biological form, with profound implications for biomedical and clinical research. Key takeaways include the critical need to address methodological challenges like measurement error and the out-of-sample problem to ensure reliable applications in fields from drug discovery to clinical malnutrition screening. Emerging directions, such as neural field representations for eigenanalysis across shape families and differentiable shape spaces, promise to further bridge geometry, physics, and design. These advances will enable more predictive virtual screening, nuanced phenotypic drug profiling, and accessible diagnostic tools, ultimately accelerating therapeutic development and improving health outcomes.
References
- 1. Morphometrics [https://en.wikipedia.org/wiki/Morphometrics]
- 2. The Importance of Discerning Shape in Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2854656/]
- 3. Geometric morphometrics and geological shape- ... [https://www.sciencedirect.com/science/article/abs/pii/S0012825202000685]
- 4. Topology: The Mathematics of Shape, Space, and Continuity [https://medium.com/prismai/topology-the-mathematics-of-shape-space-and-continuity-8c221be5c59a]
- 5. Topological space [https://en.wikipedia.org/wiki/Topological_space]
- 6. Assessing the application of landmark-free morphometrics to ... [https://bmcecolevol.biomedcentral.com/articles/10.1186/s12862-025-02377-9]
- 7. Advances in the Development of Shape Similarity Methods ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6068280/]
- 8. OptiPharm: An evolutionary algorithm to compare shape ... [https://www.nature.com/articles/s41598-018-37908-6]
- 9. Size, shape, and form: concepts of allometry in geometric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4896994/]
- 10. Walking on Kendall's Shape Space: Understanding ... [https://link.springer.com/article/10.1007/s11692-020-09513-x]
- 11. Shape Space - Morphomatics [https://morphomatics.github.io/tutorials/tutorial_shape_space/]
- 12. Probabilistic U-Net with Kendall Shape Spaces for ... [https://arxiv.org/html/2410.14017v1]
- 13. A Practical Guide to Sliding and Surface Semilandmarks in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7780474/]
- 14. A Comparison of Semilandmarking Approaches in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10093231/]
- 15. A Comparison of Semilandmarking Approaches in the ... [https://www.mdpi.com/2076-2615/13/3/385]
- 16. A novel investigation of the effect of iterations in sliding semi ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-3497-7]
- 17. Geometric morphometrics approach for classifying ... [https://www.nature.com/articles/s41598-025-85718-4]
- 18. Geometric morphometrics approach for classifying ... [https://pubmed.ncbi.nlm.nih.gov/39890869/]
- 19. "Geometric Morphometric" Approach To Detect Body ... [https://ui.adsabs.harvard.edu/abs/2025TIJMS..41..185D/abstract]
- 20. Defining, Reconstructing and Investigating Shape Through a ... [https://journal.caa-international.org/articles/10.5334/jcaa.161]
- 21. An image-computable model of human visual shape similarity [https://pmc.ncbi.nlm.nih.gov/articles/PMC8195351/]
- 22. Unlocking species identity: geometric morphometrics of head ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11931392/]
- 23. Artificial intelligence in drug discovery and development - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7577280/]
- 24. Automated morphological phenotyping using learned shape descriptors and functional maps: A novel approach to geometric morphometrics | PLOS Computational Biology [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1009061]
- 25. [2003.01357] Shape analysis via inconsistent surface registration [https://ar5iv.labs.arxiv.org/html/2003.01357]
- 26. Review of statistical shape spaces for 3D data with ... [https://www.sciencedirect.com/science/article/abs/pii/S1077314214001131]
- 27. Digital morphometrics: Application of MorphoLeaf in shape visualization and species delimitation, using Cucurbitaceae leaves as a model - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8564096/]
- 28. Convolutional neural networks and outline analyses for ... [https://peercommunityjournal.org/articles/10.24072/pcjournal.647/]
- 29. A review of machine learning applications for identification ... [https://www.sciencedirect.com/science/article/pii/S1574954125003383]
- 30. Current Trends and Future Opportunities of AI-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12564989/]
- 31. What Is Linear Discriminant Analysis? [https://www.ibm.com/think/topics/linear-discriminant-analysis]
- 32. Linear Discriminant Analysis in Machine Learning [https://www.geeksforgeeks.org/machine-learning/ml-linear-discriminant-analysis/]
- 33. Support Vector Machine (SVM) Algorithm [https://www.geeksforgeeks.org/machine-learning/support-vector-machine-algorithm/]
- 34. What Is Support Vector Machine? [https://www.ibm.com/think/topics/support-vector-machine]
- 35. 10 Pros & Cons of Support Vector Machines [2025] [https://digitaldefynd.com/IQ/pros-cons-of-support-vector-machines/]
- 36. Testing the reliability of geometric morphometric and ... [https://www.sciencedirect.com/science/article/pii/S2666033425000048]
- 37. Geometric morphometrics approach for classifying children's ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11785739/]
- 38. Comparative study of encoded and alignment-based ... [https://www.nature.com/articles/s41598-023-45461-0]
- 39. Landmark-Free Morphometric Analysis of Distal Radii [https://pmc.ncbi.nlm.nih.gov/articles/PMC11943171/]
- 40. FastROCS Shape based virtual screening software [https://www.eyesopen.com/fastrocs]
- 41. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 42. Electrostatic-field and surface-shape similarity for virtual ... [https://optibrium.com/knowledge-base/electrostatic-field-and-surface-shape-similarity-for-virtual-screening-and-pose-prediction/]
- 43. Top 5 Drug Discovery Trends 2025 Driving Breakthroughs [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 44. DeepDTAGen: a multitask deep learning framework for ... [https://www.nature.com/articles/s41467-025-59917-6]
- 45. Inside ELRIG's Drug Discovery 2025 [https://www.drugtargetreview.com/article/190237/inside-elrigs-drug-discovery-2025/]
- 46. Module: 5. Nutrition : View as single page Nutritional Assessment [https://www.open.edu/openlearncreate/mod/oucontent/view.php?id=318&printable=1]
- 47. Simple measures - arm anthropometry [https://www.measurement-toolkit.org/anthropometry/objective-methods/simple-measures-muac]
- 48. Investigation of Shape with Patients Suffering from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5754437/]
- 49. Enhancing nutritional status prediction through attention ... [https://www.sciencedirect.com/science/article/pii/S2666521225000596]
- 50. DeepSSM: A Deep Learning Framework for Statistical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6385885/]
- 51. Development and external validation of machine learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12225150/]
- 52. An algorithm for learning shape and appearance models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6554617/]
- 53. Geometric Morphometrics of Astragalus and Shape Variation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12023762/]
- 54. A Comparative Evaluation of Shape Descriptors [https://arxiv.org/html/2411.00561v1]
- 55. HusMorph: a simple machine learning app for automated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12543357/]
- 56. Noctuidae) to support pest identification in invasive species ... [https://www.frontiersin.org/journals/insect-science/articles/10.3389/finsc.2025.1542467/full]
- 57. HusMorph: A simple machine learning app for automated ... [https://ecoevorxiv.org/repository/view/8611/]
- 58. Automated image-based identification and consistent ... [https://www.sciencedirect.com/science/article/pii/S2666165925000122]
- 59. Optimizing digitalization effort in morphometrics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7723759/]
- 60. An example of inter-operator bias in 3D landmarks and its ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0197675]
- 61. Resolving sample identification bias in morphometrics ... [https://elifesciences.org/reviewed-preprints/94685v1]
- 62. Classification methods for planar shapes [https://www.sciencedirect.com/science/article/abs/pii/S0957417420301457]
- 63. Classifying high-dimensional phenotypes with ensemble ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10312448/]
- 64. 10 Machine Learning Algorithms to Know in 2025 [https://www.coursera.org/articles/machine-learning-algorithms]
- 65. 50 Statistics Every Technology Leader Should Know in 2025 [https://www.integrate.io/blog/data-transformation-challenge-statistics/]
- 66. Urban Institute Data Visualization style guide - GitHub Pages [http://urbaninstitute.github.io/graphics-styleguide/]
- 67. Understanding Re-identification Risk when Linking ... [https://privacy-analytics.com/resources/articles/managing-re-identification-risk-when-linking-multiple-datasets]
- 68. Pooling Different Safety Data Sources: Impact of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6739274/]
- 69. Diving into Best Practices for Pooling Clinical Trial Data [https://www.certara.com/blog/diving-into-best-practices-for-pooling-clinical-trial-data/]
- 70. The Hidden Dangers Of Data Pooling: How It Can Expose ... [https://littlebigtech.co.uk/the-hidden-dangers-of-data-pooling-how-it-can-expose-sensitive-information/]
- 71. Top 10 Data Security Risks In 2025 & How To Prevent Them [https://securiti.ai/data-security-risks/]
- 72. Optimal data pooling from multiple waterbodies to improve ... [https://www.sciencedirect.com/science/article/pii/S0169772225002657]
- 73. Basic Considerations for Data Pooling Strategy in Multi- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11880086/]
- 74. Classifying organisms and artefacts by their outline shapes [https://pmc.ncbi.nlm.nih.gov/articles/PMC9554513/]
- 75. Automated landmarking via multiple templates - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9714854/]
- 76. Multi-template tensor-based morphometry [https://www.sciencedirect.com/science/article/abs/pii/S1053811911003053]
- 77. Functional data geometric morphometrics with machine ... [https://www.nature.com/articles/s41598-024-66246-z]
- 78. Longitudinal deformation-based morphometry pipeline to ... [https://apertureneuro.org/article/133510-longitudinal-deformation-based-morphometry-pipeline-to-study-neuroanatomical-differences-in-structural-mri-based-on-syn-unbiased-templates]
- 79. Train-val-test splits and cross-validation [https://medium.com/@masadeghi6/how-to-split-your-data-for-machine-learning-eae893a8799c]
- 80. When Shape Defines: Geometric Morphometrics Applied to ... [https://www.mdpi.com/1424-2818/17/10/680]
- 81. Cross Validation: Training, Validation and Testing Splits [https://the-examples-book.com/tools/data-modeling-cross-validation-train-valid-test]
- 82. 5.1. Cross-validation — Transparent ML Intro [https://alan-turing-institute.github.io/Intro-to-transparent-ML-course/05-cross-val-bootstrap/cross-validation.html]
- 83. 3.1. Cross-validation: evaluating estimator performance [https://scikit-learn.org/stable/modules/cross_validation.html]
- 84. Cross Validation in Machine Learning [https://www.geeksforgeeks.org/machine-learning/cross-validation-machine-learning/]
- 85. Data splits and cross-validation in automated machine ... [https://learn.microsoft.com/en-us/azure/machine-learning/how-to-configure-cross-validation-data-splits?view=azureml-api-1]
- 86. Geometric morphometric analysis of body shape and ... [https://www.sciencedirect.com/science/article/abs/pii/S0940960225002869]
- 87. Deep Learning Outperforms Geometric Morphometrics for ... [https://archaeo.peercommunityin.org/PCIArchaeology/articles/rec?id=502]
- 88. Symmetry in Explainable AI: A Morphometric Deep ... [https://www.mdpi.com/2073-8994/17/8/1264]
- 89. An artificial intelligence accelerated virtual screening ... [https://www.nature.com/articles/s41467-024-52061-7]
- 90. A Geometric Morphometric Study of Scapular Ontogeny in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12236271/]
- 91. Retrospective Validation [https://kneat.com/glossary/retrospective-validation/]
- 92. Retrospective Validation in Legacy Systems [https://pharmuni.com/glossary/retrospective-validation/]
- 93. An Improved Metric and Benchmark for Assessing the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10980085/]
- 94. Benchmarking matters | Nature Biomedical Engineering [https://www.nature.com/articles/s41551-025-01572-9]
- 95. 2025 search tool benchmark: key metrics to evaluate ... [https://www.glean.com/blog/2025-search-tool-benchmark-key-metrics-to-evaluate-accuracy-and-speed]
- 96. Applying machine learning to assist in the morphometric ... [https://pubmed.ncbi.nlm.nih.gov/40510864/]
- 97. 20 Best Performance Testing Tools to Use in 2025 [https://www.headspin.io/blog/best-performance-testing-tools]
- 98. Comprehensive benchmarking of computational tools for ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00931-z]
- 99. Machine learning models for predicting morphological ... [https://www.nature.com/articles/s41598-025-15373-2]
- 100. Advances in deep learning for cellular dynamics analysis [https://www.sciencedirect.com/science/article/pii/S0955067425001231]
- 101. Comparing JMeter with Other Performance Testing Tools in ... [https://www.frugaltesting.com/blog/comparing-jmeter-with-other-performance-testing-tools-in-2025]
- 102. Enhancing Reproducibility in Biomedical Research ... [https://www.nationalacademies.org/our-work/enhancing-reproducibility-in-biomedical-research-through-harmonization-of-guidelines-for-transparent-reporting-a-workshop]
- 103. On the use of shape spaces to compare morphometric ... [http://www.italian-journal-of-mammalogy.it/On-the-use-of-shape-spaces-to-compare-morphometric-methods,77659,0,2.html]
- 104. Presenting data in tables and charts - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4008059/]
- 105. Presenting Quantitative Data Graphically [https://courses.lumenlearning.com/waymakermath4libarts/chapter/presenting-quantitative-data-graphically/]
- 106. Presentation of Quantitative Data [https://ihatepsm.com/blog/presentation-quantitative-data]