Phenotypic vs. Target-Based Screening for Antimalarials: A Strategic Guide for Drug Discovery
With artemisinin resistance threatening global malaria control, the need for novel antimalarials is more urgent than ever.
Phenotypic vs. Target-Based Screening for Antimalarials: A Strategic Guide for Drug Discovery
Abstract
With artemisinin resistance threatening global malaria control, the need for novel antimalarials is more urgent than ever. This article provides a comprehensive comparison of the two principal drug discovery paradigms—phenotypic and target-based screening—in the context of antimalarial development. Tailored for researchers and drug development professionals, we explore the foundational principles, methodological applications, and inherent challenges of each approach. The review highlights how phenotypic screening has been the dominant source of recent clinical candidates by enabling target-agnostic discovery of first-in-class medicines. Concurrently, we examine the resurgence of target-based strategies, fueled by new technologies for target deconvolution and validation. By synthesizing recent successes, troubleshooting common pitfalls, and presenting a comparative framework, this article serves as a strategic guide for selecting and optimizing screening methodologies to accelerate the pipeline of next-generation antimalarial therapies.
Malaria's Urgent Threat and the Foundations of Drug Discovery
The Global Burden of Malaria and the Challenge of Drug Resistance
Malaria remains one of the most devastating parasitic infectious diseases, with an estimated 263 million cases and approximately 597,000 deaths annually, primarily affecting low- and middle-income countries [1] [2]. The fight against malaria is severely challenged by the relentless emergence and spread of drug-resistant Plasmodium falciparum parasites, the deadliest species causing the disease. Artemisinin partial resistance (ART-R), characterized by delayed parasite clearance after treatment, has now been confirmed in Southeast Asia and, alarmingly, has recently emerged in East Africa [3]. This evolution of resistance undermines the efficacy of frontline artemisinin-based combination therapies (ACTs), threatening to reverse decades of progress in malaria control. In this context, the discovery of new antimalarials with novel mechanisms of action is a global health priority. This guide compares the two principal drug discovery approaches—phenotypic screening and target-based screening—evaluating their performance and utility in antimalarial research.
Table 1: Fundamental Comparison of Screening Approaches
| Feature | Phenotypic Screening | Target-Based Screening |
|---|---|---|
| Basic Principle | Measures compound's effect on whole parasites (cells) in culture [4]. | Measures compound's effect on a specific, purified molecular target [4]. |
| Key Advantage | Identifies compounds permeable to cell membrane; can discover compounds with novel or polypharmacological mechanisms [4]. | Allows for rational, target-focused design; typically has a straightforward mechanistic output [4]. |
| Hit Identification | Directly identifies compounds that kill the parasite or inhibit its growth. | Identifies compounds that inhibit the function of the specific target. |
| Target Deconvolution | Required after hit identification; can be complex and time-consuming [5]. | Known from the outset of the screen. |
| Success Rate | Historically more successful for discovering first-in-class antimalarial small molecules [6]. | Has contributed fewer new chemical entities to the antimalarial pipeline [6]. |
Phenotypic Screening: A Direct Path to Parasite Killers
Phenotypic screening involves testing compounds for their ability to kill or inhibit the growth of the entire parasite in its cellular context. This approach has been the most productive method for discovering new antimalarial leads over the past decade [6].
Experimental Protocol: Image-Based Antimalarial High-Throughput Screening (HTS)
A contemporary protocol for phenotypic HTS against the asexual blood stage of P. falciparum involves the following key steps [1]:
- Compound Library Preparation: An in-house library of thousands of small molecules is prepared in stock solutions, often in dimethyl sulfoxide (DMSO), and dispensed into multi-well plates (e.g., 384-well format).
- Parasite Culture & Synchronization: P. falciparum parasites (including drug-sensitive and resistant strains like 3D7, K1, Dd2) are cultured in human red blood cells (RBCs) and synchronized at the desired developmental stage using agents like sorbitol.
- Compound Exposure & Incubation: The synchronized parasite culture is dispensed into the compound-treated plates and incubated for a set period (e.g., 72 hours) under controlled atmospheric conditions (e.g., 1% O₂, 5% CO₂ in N₂ at 37°C).
- Staining & Image Acquisition: After incubation, the plates are stained with fluorescent dyes. A common combination includes:
- Wheat germ agglutinin–Alexa Fluor 488: Stains the RBC membrane.
- Hoechst 33342: Stains parasite nucleic acid. The plates are then fixed (e.g., with paraformaldehyde) and imaged using high-content imaging systems (e.g., Operetta CLS).
- Image Analysis: Automated image analysis software (e.g., Columbus) is used to quantify parasite viability and growth based on the fluorescence signals.
Key Discoveries from Phenotypic Screening
This approach has yielded numerous promising compounds with multi-stage activity, meaning they are effective against more than one life cycle stage of the parasite [5].
Table 2: Recent Multi-Stage Antimalarial Hits from Phenotypic Screens
| Compound | Primary Molecular Target / Mechanism | Asexual Blood Stage (TCP-1) | Liver Stage (TCP-4) | Sexual Stage / Transmission (TCP-5) |
|---|---|---|---|---|
| MMV030084 | Plasmodium cGMP-dependent protein kinase (PfPKG) inhibitor; disrupts merozoite egress [5]. | Yes (nanomolar potency) | Yes (vs. P. berghei) | Yes (inhibits gametocyte activation) |
| WM382 | Dual inhibitor of Plasmepsin IX (PMIX) and X (PMX); prevents merozoite egress and invasion [5]. | Yes (nanomolar potency, no cross-resistance) | Yes (reduces merozoite viability) | Yes (inhibits oocyst development) |
| SGI-1027 | DNA methyltransferase (DNMT) inhibitor; disrupts epigenetic regulation [5]. | Yes (low-nanomolar potency) | Information missing from search results | Yes (vs. early-stage gametocytes) |
Table abbreviations: TCP, Target Candidate Profile.
Target-Based Screening: A Focused, Rational Approach
In contrast, target-based screening begins with a specific, validated molecular target—typically a protein enzyme or receptor essential for parasite survival. Compounds are then screened for their ability to modulate the activity of this purified target.
Experimental Protocol: In Vitro Enzyme Assay
A generalized protocol for a target-based HTS is as follows:
- Target Selection & Production: A recombinant form of the target protein (e.g., PfDHFR, PfPKG, PfATP4) is expressed and purified.
- Assay Development: A biochemical assay is optimized to measure the target's activity. This often uses a fluorescent or luminescent readout to monitor substrate conversion or product formation in a plate-based format.
- Compound Screening: The purified target is exposed to the compound library, and its activity is measured. Compounds that significantly inhibit the activity are identified as "hits."
- Cellular Validation: Hits from the primary screen must then be tested in whole-parasite assays to confirm they can cross the parasite membranes and exert the intended antimalarial effect.
Performance Comparison: Data and Outcomes
The two strategies can be evaluated based on their empirical success in delivering new antimalarial candidates and their operational characteristics.
Quantitative Success in Lead Generation
Large-scale phenotypic screening campaigns have been a major source of new leads. For instance, a high-throughput screen of a 9,547-compound library identified 256 active compounds (top 3%), with 110 of these being novel and 157 exhibiting high potency (IC₅₀ < 1 µM) [1]. Another major screen of over 2 million compounds from the GSK corporate collection identified 13,533 phenotypic hits [6]. This wealth of starting points has fed the drug development pipeline, with several candidates progressing to clinical trials.
Operational and Strategic Comparison
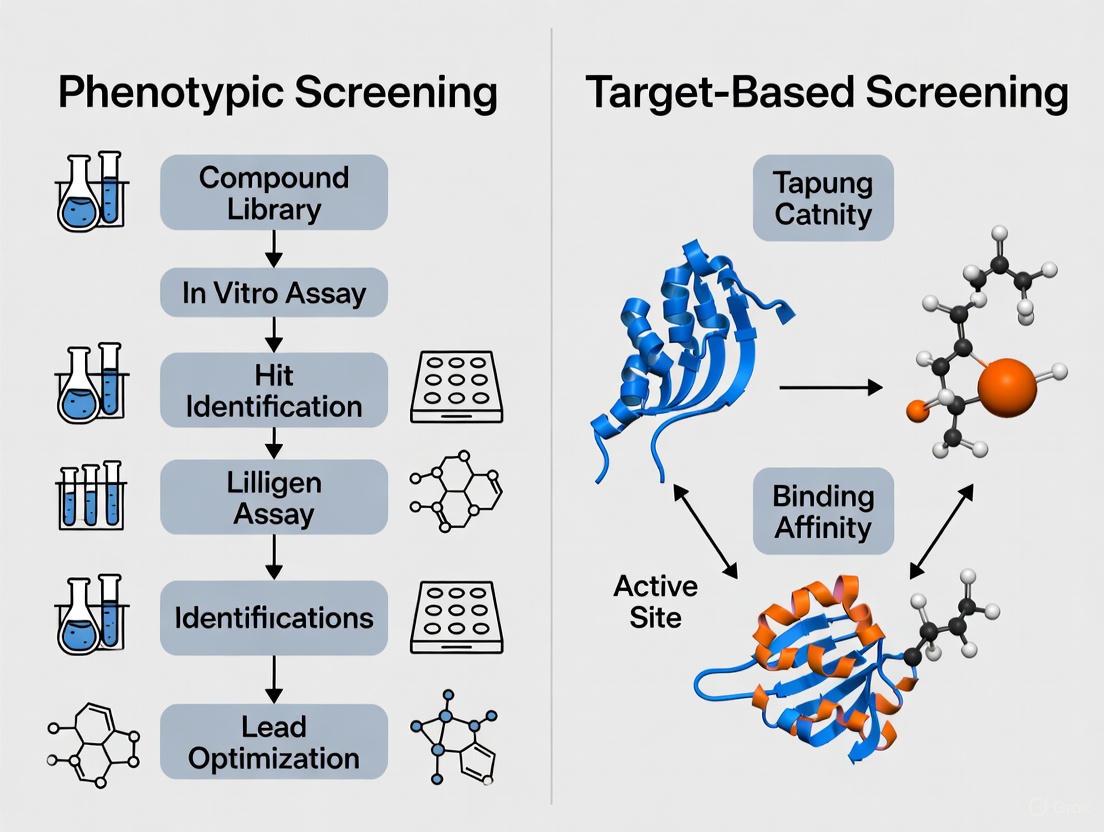
The following diagram illustrates the core workflows and key differentiators of each screening approach.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of both screening strategies relies on a suite of critical reagents and tools.
Table 3: Key Research Reagent Solutions for Antimalarial Screening
| Reagent / Tool | Function in Research | Application Example |
|---|---|---|
| Synchronized P. falciparum Cultures | Provides developmentally staged parasites for reproducible screening. | Essential for phenotypic HTS to ensure consistent compound exposure across parasite stages [1]. |
| Fluorescent Nucleic Acid Stains (e.g., Hoechst 33342, SYBR Green) | Enables quantification of parasite growth and viability in high-throughput formats. | Used in image-based phenotypic screens to stain parasite DNA for automated quantification [1] [4]. |
| Recombinant Parasite Proteins (e.g., PfK13, PfPKG, PfDHFR) | Serves as the defined molecular target for biochemical inhibition assays. | Critical for target-based screens and for validating the mechanism of action of hits from phenotypic screens [3] [5]. |
| Genetically Modified Parasite Strains | Allows for the study of resistance mechanisms and target validation. | Parasites with mutations in kelch13 are used to validate ART-R and screen for compounds effective against resistant strains [1] [3]. |
Both phenotypic and target-based screening are indispensable tools in the antimalarial research arsenal. Phenotypic screening has proven highly effective in delivering novel, potent, and multi-stage antimalarial leads with a higher historical success rate for discovering first-in-class drugs. Its key strength lies in its ability to identify compounds that are inherently active against the whole parasite, often revealing novel biology. Target-based screening offers a more focused path with a clear mechanistic hypothesis from the start, facilitating rational drug optimization. The future of antimalarial discovery lies in leveraging the strengths of both approaches—using phenotypic screening to identify potent, novel chemotypes, and employing target-based methods to optimize hits and ensure a clear resistance management strategy—in order to outpace the evolving threat of drug-resistant malaria.
In the relentless fight against malaria, a disease that continues to cause major mortality globally, drug discovery hinges on two principal screening methodologies: phenotypic screening and target-based screening [7]. Over the past two decades (2005-2025), the antimalarial development pipeline has been significantly shaped by these two philosophies [7]. Phenotypic screening evaluates the overall biological effect of a compound on whole parasite cells, observing changes in the parasite's phenotype without prior assumptions about the molecular target [8] [4]. In contrast, target-based screening focuses on identifying compounds that interact with a specific, purified molecular target believed to be critical to the parasite's survival [1] [8]. This guide provides an objective comparison of these two paradigms, detailing their core principles, experimental protocols, and performance within the context of antimalarial research.
Conceptual Comparison: Phenotypic vs. Target-Based Screening
The strategic dichotomy between these approaches lies in their starting point and underlying philosophy. The following diagram illustrates the fundamental workflows of each paradigm.
The core distinction is that phenotypic screening begins with a holistic biological system, while target-based screening begins with a specific, isolated molecular component. Phenotypic screening does not rely on prior knowledge of the disease's molecular underpinnings, capturing potential therapeutic benefits that may not be tied to a single molecular interaction [8]. Conversely, the target-based approach requires a deep understanding of the disease’s molecular mechanisms and precise validation of the chosen target [8].
Performance and Outcome Analysis
The ultimate measure of a screening strategy is its success in delivering clinical candidates. Analysis of antimalarial drug development over the last two decades reveals a clear trend.
Table 1: Comparative Success in Antimalarial Drug Discovery (2005-2025)
| Screening Paradigm | Key Advantages | Key Limitations | Notable Antimalarial Successes | Relative Success Rate in Delivering Clinical Candidates |
|---|---|---|---|---|
| Phenotypic Screening | • Identifies first-in-class drugs [4]• Does not require prior target knowledge [8]• Accounts for cell permeability & metabolism [4]• Can uncover novel mechanisms of action [4] | • Mechanism of action initially unknown [8]• Can be more resource-intensive [8]• Target deconvolution can be challenging | • Artemisinin (via traditional approach) [8]• Spiroindolone KAE609 (in clinical trials) [4] | Higher - Outperformed target-based approach in this period [7] |
| Target-Based Screening | • High throughput and cost-effective [8]• Allows for precise drug optimization [8]• Mechanism of action is known from the start | • Only as good as the underlying disease knowledge [8]• Requires a validated, druggable target• May not account for cell permeability | • HIV antiretroviral therapies (e.g., Raltegravir) [8] | Lower - Underperformed compared to phenotypic approach in antimalarials [7] |
Recent analyses confirm that phenotypic-based drug discovery has outperformed the target-based approach in populating the antimalarial clinical pipeline [7]. This is largely attributed to the complex biology of the Plasmodium parasite, where many pathways are still not fully understood, making the unbiased nature of phenotypic screening particularly advantageous [8] [4].
Experimental Protocols and Data Output
To illustrate the practical application of these paradigms, below are detailed methodologies from recent, high-throughput research.
Detailed Protocol: Phenotypic High-Throughput Screening
This protocol, adapted from a 2025 study, describes an image-based phenotypic screen to identify novel antimalarial agents [1].
- Objective: To identify novel antimalarial compounds through their effect on the viability of whole Plasmodium falciparum parasites.
- Parasite Strains: CQ-sensitive (3D7, NF54) and CQ-resistant (K1, Dd2) strains, among others [1].
- Compound Library: An in-house library of 9,547 small molecules [1].
Workflow:
- Culture & Synchronization: P. falciparum parasites are maintained in human O+ red blood cells (RBCs) and double-synchronized at the ring stage using 5% sorbitol treatment [1].
- Compound Dispensing: Library compounds are arrayed in 384-well plates at a single concentration (e.g., 10 µM) or in a dose-dependent manner. Chloroquine is used as a control [1].
- Inoculation & Incubation: Synchronized parasite cultures (1% schizont-stage at 2% haematocrit) are dispensed into compound-treated plates and incubated for 72 hours [1].
- Staining & Imaging: After incubation, the plate is stained with a solution containing:
- Wheat agglutinin–Alexa Fluor 488: Stains the RBC membrane.
- Hoechst 33342: Stains parasitic nucleic acid.
- Paraformaldehyde: Fixes the culture.
- Image Acquisition & Analysis: Nine image fields per well are acquired using a high-content imaging system (e.g., Operetta CLS). Automated image analysis software (e.g., Columbus) is used to classify parasites and quantify viability based on the fluorescent signals [1].
Table 2: Key Reagents for Phenotypic Antimalarial Screening
| Research Reagent | Function in the Experiment |
|---|---|
| Synchronized P. falciparum Cultures | Provides the biological system for evaluating compound effects on the entire parasite lifecycle stage. |
| In-house Compound Library | The source of potential drug candidates being tested for biological activity. |
| Wheat agglutinin–Alexa Fluor 488 | A fluorescent dye that binds to the red blood cell membrane, enabling visualization of host cells. |
| Hoechst 33342 | A nucleic acid intercalating dye that specifically stains parasite DNA, allowing for quantification of parasite load and viability. |
| High-Content Imaging System | Enables automated, high-throughput capture of fluorescent images for quantitative analysis of phenotypic changes. |
Detailed Protocol: A Hybrid Apicoplast Screening Approach
Some screens represent a hybrid strategy, starting as phenotypic but being tailored to a specific biological process. A prime example is the search for apicoplast-targeting compounds [4].
- Objective: To identify compounds that target the apicoplast organelle, which produces a distinctive "delayed death" phenotype.
- Rationale: Drugs that disrupt essential housekeeping functions in the apicoplast allow parasites to complete one 48-hour lifecycle, but their progeny fail to develop and die [4].
Workflow:
- Phenotypic Assay: A standard luciferase-based parasite viability assay is performed.
- Dual Time-Point Measurement: The effect of compounds is measured at both 48 hours and 96 hours.
- Hit Identification: Compounds that show significantly higher potency at the 96-hour time point are flagged as potential apicoplast-targeting agents, as they induce the "delayed death" effect [4].
The following diagram visualizes this specific screening strategy and the unique phenotype it detects.
Quantitative Data from Screening Campaigns
The output of a high-throughput screen is a rich dataset used to prioritize hits for further development. The 2025 HTS and meta-analysis study provides a clear example of the hit confirmation and triage process [1].
Table 3: Hit Triage Data from a Phenotypic HTS Campaign
| Selection Criteria | Number of Compounds | Purpose of Selection |
|---|---|---|
| Primary HTS Hits (Top 3%) | 256 | Initial actives selected from 9,547 compounds for dose-response analysis [1]. |
| Confirmed Activity (IC₅₀ < 1 µM) | 157 | Confirmation of potent antimalarial activity in a dose-dependent manner [1]. |
| Novel & Unpublished | 110 | Focus on new chemical space and novel mechanisms of action [1]. |
| Favorable Safety (LD₅₀/MTD > 20 mg/kg) | 69 | Early removal of compounds with potential toxicity issues [1]. |
| Final In Vivo Candidates | 3 | Potent inhibitors showing suppression in a P. berghei mouse model [1]. |
This data demonstrates the rigorous filtering process required to move from thousands of initial screening hits to a handful of viable lead candidates, a process critical for both phenotypic and target-based approaches.
Both phenotypic and target-based screening are indispensable tools in modern antimalarial drug discovery. The empirical evidence from the last two decades shows that the phenotypic approach has been more successful in delivering clinical candidates, largely due to its ability to identify novel chemotypes without being limited by current, and often incomplete, biological knowledge [7] [4]. However, the target-based approach offers precision and efficiency that are invaluable once a target is validated [8]. The future of antimalarial research lies not in choosing one paradigm over the other, but in strategically employing them as complementary forces. Emerging hybrid strategies, like the apicoplast screen, alongside advanced technologies such as AI-integrated platforms [9] and PBPK modeling [10] [11], will further enhance our ability to discover the next generation of life-saving malaria treatments.
The relentless fight against malaria, a disease causing an estimated 409,000 deaths annually, is critically dependent on the continuous pipeline of new therapeutic compounds [12]. The emergence and spread of artemisinin resistance threaten the efficacy of current first-line treatments, making the discovery of novel antimalarials more urgent than ever [7] [13]. Within this high-stakes context, antimalarial drug discovery is primarily guided by two distinct screening philosophies: phenotypic screening and target-based screening [7] [4]. This guide provides a detailed, objective comparison of these paradigms, with a focused examination of the core principles, experimental protocols, and practical applications of target-based screening. Understanding this strategic dichotomy is essential for research teams to allocate resources effectively and accelerate the development of new medicines capable of combating drug-resistant Plasmodium parasites [8].
Core Philosophical and Practical Distinctions
At its core, the distinction between phenotypic and target-based screening lies in the fundamental starting point of the discovery process.
Phenotypic Screening: This is a holistic approach that assesses the overall effect of a compound on whole cells or entire organisms without prior knowledge of a specific molecular target. In malaria research, this involves screening compounds against live Plasmodium falciparum parasites cultured in human red blood cells [14] [4]. The primary advantage is its ability to identify compounds that effectively kill the parasite through novel or multiple mechanisms of action (polypharmacology), even when the underlying biology is poorly understood [8] [4]. Historically, this approach has outperformed target-based methods in delivering new clinical candidates for malaria [7]. The discovery of artemisinin itself is a classic example of a phenotypic success, where the potent antimalarial effect was identified before its molecular target was fully elucidated [8].
Target-Based Screening: This is a reductionist strategy that begins with a specific, validated macromolecule—typically a protein—known to be essential for parasite survival or pathogenesis. The goal is to identify compounds that selectively interact with and modulate the activity of this predefined target [12] [8]. This approach requires a deep understanding of parasite biology to select a "druggable" target. Its strengths include precision, the ability to employ sophisticated methods like fragment-based screening and structure-based drug design, and the capacity to optimize for selectivity against human orthologs early in the process [12].
Table 1: High-Level Comparison of Screening Paradigms in Antimalarial Research
| Feature | Phenotypic Screening | Target-Based Screening |
|---|---|---|
| Starting Point | Whole parasite (e.g., blood-stage parasites in RBCs) [14] | Purified protein or validated molecular target [12] |
| Knowledge Prerequisite | Not required; can work with incomplete biology [4] | Required; depends on prior target validation [12] |
| Key Advantage | Identifies compounds with cellular activity; captures polypharmacology [4] | Enables rational, structure-based optimization and mechanistic clarity [12] |
| Primary Challenge | Target deconvolution can be difficult and resource-intensive [12] | Biochemical hit may not penetrate the parasite or kill it effectively [12] |
| Throughput Potential | Very High (e.g., image-based ultra-HTS of millions of compounds) [14] [4] | Extremely High (e.g., biochemical assays for purified enzymes) [8] |
| Impact on Pipeline | Has delivered more clinical candidates in the last two decades [7] | Potential to identify novel chemotypes with optimized properties [12] [15] |
The Target-Based Screening Workflow: Principles in Practice
The implementation of target-based screening follows a structured, multi-stage workflow. The diagram below outlines the key steps from initial target selection to hit identification.
Core Principle 1: Target Selection and Validation
The foundation of a successful target-based campaign is the selection of a validated, essential, and druggable molecular target. Criteria for prioritization include [12]:
- Essentiality: The target must be critical for parasite survival, replication, or transmission. Genetic validation (e.g., gene knockouts) is a key source of evidence.
- Druggability: The target should possess a binding site that can be potently and selectively targeted by a small molecule.
- Selectivity: The target should be sufficiently different from its human ortholog to minimize host toxicity.
- Tractability: The target must be expressible and stable in a format suitable for high-throughput screening.
In antimalarial research, targets are often identified through target deconvolution of phenotypic hits. Techniques like in vitro resistance generation followed by whole-genome sequencing or thermal proteome profiling are used to find the protein target of a compound that shows antimalarial activity [12]. Other sources include fundamental research and literature precedent. A prominent example is falcipain-2 (FP2), a cysteine protease essential for hemoglobin degradation in the parasite's digestive vacuole, making it a well-validated target for drug discovery [15].
Core Principle 2: Assay Development and High-Throughput Screening
Once a target is selected, a robust biochemical assay is developed to measure its activity in the presence of test compounds. For an enzyme like FP2, this typically involves monitoring the cleavage of a synthetic peptide substrate [15].
- Example Protocol: Biochemical Inhibition Assay for Falcipain-2
- Reagent Preparation: Purified recombinant FP2 protein is activated in an appropriate buffer. A fluorogenic substrate (e.g., Z-Leu-Arg-AMC) is prepared. Test compounds are serially diluted in DMSO [15].
- Assay Execution: In a microtiter plate, compounds are incubated with the activated enzyme. The reaction is initiated by adding the substrate. The increase in fluorescence from the cleaved product is measured over time using a plate reader [15].
- Data Analysis: The percentage of enzyme inhibition at each compound concentration is calculated. Dose-response curves are generated to determine the half-maximal inhibitory concentration (IC₅₀), a key quantitative measure of compound potency [15].
This biochemical assay format is easily miniaturized and automated, allowing for the high-throughput screening (HTS) of hundreds of thousands of compounds in a short time [8]. Furthermore, the known target structure enables target-based virtual screening (TBVS), a computational method that uses molecular docking to rapidly prioritize compounds from large digital libraries (e.g., the ZINC database) that are predicted to bind favorably to the target, such as FP2, before any wet-lab testing [15].
Core Principle 3: Hit Validation and Progression
Compounds identified from the primary screen ("hits") must be rigorously validated to ensure their activity is genuine and meaningful.
- Secondary Assays: Hits are retested in dose-response to confirm potency (IC₅₀). Counter-screens are run against related human proteases (e.g., human cathepsins) to assess selectivity and minimize off-target effects [15].
- Cellular Activity: A critical step is to demonstrate that the target inhibitor also kills the whole parasite. The compound is tested against cultured Plasmodium falciparum in vitro to determine its half-maximal effective concentration (EC₅₀). A significant disconnect between a potent biochemical IC₅₀ and a weak cellular EC₅₀ can indicate poor membrane permeability or efflux by the parasite [12].
- Cytotoxicity: The compound is tested against mammalian cell lines (e.g., human HepG2 cells) to establish a preliminary safety profile and calculate a selectivity index [15].
Table 2: Quantitative Data from a Target-Based Campaign against Falcipain-2 [15]
| Compound / Parameter | FP2 Biochemical IC₅₀ | P. falciparum (3D7) EC₅₀ | Human Cell Cytotoxicity (CC₅₀) | Selectivity Index (SI) |
|---|---|---|---|---|
| ST72 (ZINC12900664) | Not Specified (Potent binder) | 2.8 µM | > 100 µM | > 35.7 |
| Reference CQS Strain | N/A | N/A | N/A | N/A |
| Reference CQR Strain | N/A | 6.7 µM (RKL-9) | > 100 µM | > 14.9 |
The Scientist's Toolkit: Essential Reagents for Target-Based Screening
The following table details key reagents and materials required to establish a target-based screening platform for an enzymatic target like falcipain-2.
Table 3: Research Reagent Solutions for Target-Based Antimalarial Screening
| Reagent / Material | Function and Importance in the Screening Process |
|---|---|
| Purified Recombinant Protein | The core reagent. Requires high-purity, active protein (e.g., FP2) for biochemical assays. Often produced in E. coli or other expression systems [15]. |
| Chemical Compound Libraries | Diverse collections of small molecules (e.g., natural product libraries, synthetic compounds) used in HTS to identify initial hits [12] [15]. |
| Validated Assay Kits | Commercial kits providing optimized buffers, substrates, and controls for specific enzyme classes, which help standardize and accelerate assay development. |
| Fluorogenic/Cromogenic Substrates | Enzyme-specific peptides linked to reporter molecules (e.g., AMC). Cleavage by the active enzyme generates a detectable signal for inhibition monitoring [15]. |
| High-Throughput Screening Instrumentation | Automated liquid handlers, multi-mode plate readers (for fluorescence, absorbance), and robotic systems are essential for testing thousands of compounds rapidly and accurately [14]. |
| Virtual Screening Software | Computational tools (e.g., molecular docking software like InstaDock) for in silico prediction of compound-target interactions, enabling pre-screening of vast virtual libraries [15]. |
Target-based screening offers a powerful, rational approach to antimalarial drug discovery, characterized by its precision, suitability for modern cheminformatics methods, and the mechanistic clarity it provides for downstream optimization [12] [15]. However, its success is contingent on a deep and accurate understanding of parasite biology and target essentiality. The historical data showing that phenotypic screening has delivered more clinical candidates underscores the challenge of translating biochemical potency into whole-parasite killing [7] [12].
The future of antimalarial discovery does not lie in choosing one paradigm over the other, but in their strategic integration. Phenotypic screening can identify novel chemical starting points with whole-cell efficacy, while target-based methods provide a pathway to rationally optimize those hits once their target is deconvoluted [12] [8]. Furthermore, target-based virtual screening serves as an efficient filter to prioritize compounds for more resource-intensive phenotypic assays. By leveraging the complementary strengths of both philosophies, the scientific community can build a more robust and innovative pipeline to outpace drug resistance and achieve the ultimate goal of malaria eradication.
The complex life cycle of the Plasmodium parasite presents both a challenge and an opportunity for antimalarial drug discovery. Each distinct biological stage offers a potential point of attack for chemotherapeutic intervention. Target Candidate Profiles (TCPs) are formal definitions of the desired properties of new antimalarial compounds, and they are intrinsically linked to specific parasite stages [16] [12]. The strategy of malaria eradication, as opposed to mere control, demands drugs that go beyond simply treating symptoms. This requires compounds that can prevent transmission, protect vulnerable populations, and achieve radical cure, objectives that can only be met by targeting under-exploited phases of the parasite's development [12]. This guide examines how the biological features of each lifecycle stage directly inform the construction of TCPs, providing a structured framework for researchers aiming to develop the next generation of antimalarial medicines.
Decoding the Plasmodium Lifecycle: From Infection to Transmission
The malaria parasite's life cycle involves two hosts—a human and a female Anopheles mosquito—and encompasses several morphological and functional transformations [17] [18]. The following diagram illustrates the key stages and highlights where different TCPs are designed to interrupt the process.
Lifecycle Stage Characteristics and Corresponding TCPs
| Lifecycle Stage | Key Biological Features | Clinical/Disease Impact | Corresponding TCP |
|---|---|---|---|
| Liver Stage (Pre-erythrocytic) | Sporozoites invade hepatocytes; develop into schizonts releasing merozoites; dormant hypnozoites in P. vivax and P. ovale [17] [18]. | Establishes infection; hypnozoites cause relapses weeks or years later [17] [12]. | TCP-4: Prophylaxis (targets liver schizonts) [16] [12].TCP-3: Anti-relapse (targets hypnozoites) [16] [12]. |
| Asexual Blood Stage | Merozoites invade erythrocytes; cyclic asexual replication (schizogony); 48-72 hour cycles cause febrile paroxysms [17] [18]. | Responsible for all clinical symptoms of malaria; can progress to severe disease and death [17]. | TCP-1: Clears asexual blood-stage parasitemia; treats disease [16] [12]. |
| Sexual Stage (Gametocytes) | Differentiate in blood; male and female gametocytes are ingested by a mosquito [17]. | No clinical symptoms; essential for transmission to mosquitoes [17]. | TCP-5: Blocks transmission by killing gametocytes [16] [12]. |
Drug Discovery Strategies: Phenotypic vs. Target-Based Screening
The two primary approaches for identifying new antimalarial compounds are phenotypic screening and target-based screening. Each has distinct advantages, challenges, and is particularly suited to different stages of the parasite's life cycle and TCP goals.
Core Methodologies and Workflows
The fundamental workflows for phenotypic and target-based screening differ significantly, as illustrated below.
Phenotypic Screening involves testing compounds for their ability to induce a desired effect—such as killing parasites within human red blood cells—without prior knowledge of the specific molecular target [19] [20]. A key strength of this approach is its ability to identify compounds that are inherently cell-active and can penetrate the complex cellular environment of the parasite [19]. Its major challenge is target deconvolution—the subsequent, often difficult process of identifying the precise protein or mechanism through which the compound acts [12] [20]. Techniques for target deconvolution include generating drug-resistant parasites followed by whole-genome sequencing (WGS) to identify mutations, as well as methods like Thermal Proteome Profiling and metabolomics [12].
Target-Based Screening begins with a known, validated molecular target believed to be essential for parasite survival or transmission. Compounds are screened in biochemical assays for activity against this purified target (e.g., an enzyme) [12] [19]. The primary advantage is knowing the mechanism from the outset, which can facilitate downstream optimization and resistance monitoring. The major challenge is that a compound active in a biochemical assay may fail to be effective in a cellular or whole-parasite context due to issues with permeability, metabolism, or efflux [12].
Comparative Analysis: Applications and Limitations for Antimalarial Discovery
The choice between phenotypic and target-based screening is often guided by the specific TCP and the biological context of the target lifecycle stage.
Table 2: Comparative Analysis of Drug Discovery Screening Approaches
| Aspect | Phenotypic Screening | Target-Based Screening |
|---|---|---|
| Primary Screening Context | Whole parasites in cultured human cells (e.g., erythrocytes, hepatocytes) [12]. | Purified protein or enzyme in a biochemical assay [12]. |
| Hit Identification Basis | Measured phenotypic outcome (e.g., inhibition of parasite growth or development) [19]. | Biochemical interaction with a predefined molecular target [19]. |
| Advantages | - Unbiased; can discover novel biology/MoAs [21] [19].- Identifies cell-active compounds upfront [19].- Historically more successful for first-in-class drugs [21]. | - High throughput; can screen vast compound libraries [19].- Known target simplifies lead optimization and SAR [12].- Enables fragment-based and structure-based design [12]. |
| Key Challenges | - Target deconvolution can be complex and time-consuming [12] [20].- Lower throughput compared to target-based [19]. | - Cellular activity not guaranteed (lack of translation) [12].- Requires a priori target selection and validation [12]. |
| Relevance to TCPs | Ideal for TCP-1 (blood stage) where high-content parasite growth assays are established. Also valuable for TCP-5 (gametocyte) and liver stage (TCP-3/4) assays [12]. | Powerful for well-validated targets like PfATP4 (TCP-1) [22]. Structure-based design is crucial for overcoming resistance in known targets. |
The Scientist's Toolkit: Essential Reagents and Protocols
Advancing antimalarial drug discovery requires a suite of specialized reagents and standardized protocols tailored to interrogate different parasite stages and TCPs.
Key Research Reagent Solutions
Table 3: Essential Research Reagents for Antimalarial Discovery
| Reagent / Assay System | Primary Function in Research | Relevance to TCP |
|---|---|---|
| In vitro P. falciparum cultures (Asexual blood stages) | Platform for phenotypic screening of compound libraries for anti-parasitic activity [12]. | TCP-1: Primary screen for blood-stage clearance. |
| In vitro gametocyte cultures | Enable screening for compounds that kill or disable transmission-competent sexual stages [12]. | TCP-5: Screen for transmission-blocking activity. |
| Hepatocyte cell lines & primary hepatocytes | Model liver-stage infection for screening compounds against liver schizonts and hypnozoites [12]. | TCP-4 (prophylaxis) and TCP-3 (anti-relapse). |
| Recombinant Plasmodium proteins (e.g., PfATP4, aminoacyl-tRNA synthetases) | Targets for biochemical high-throughput screening (HTS) and structure-based drug design [12] [22]. | TCP-1: Validated targets for blood stage. |
| Transgenic parasite lines (e.g., luciferase/reporter strains) | Enable high-throughput, automated readouts of parasite growth or specific pathway inhibition [12]. | All TCPs: Facilitates compound screening across stages. |
| Animal models of malaria (e.g., P. berghei, P. cynomolgi, humanized mice) | Evaluate efficacy of lead compounds in a whole-body system, including pharmacokinetics and relapse models [12]. | All TCPs: Critical for in vivo validation. |
Detailed Experimental Protocols
To ensure reproducibility and accurate comparison of data across different laboratories, the following standardized protocols are widely used in the field.
Protocol 1: Phenotypic High-Throughput Screening for TCP-1 (Asexual Blood Stage)
- Parasite Culture: Maintain synchronized cultures of P. falciparum (e.g., strain 3D7 or Dd2) in human O+ erythrocytes at 2% hematocrit in complete RPMI 1640 medium [12].
- Compound Incubation: Transfer parasites at ~1% parasitemia (ring stage) to 384-well assay plates. Add test compounds from a library over a concentration range (e.g., 10 μM to 1 nM). Include controls (e.g., artemisinin for 100% inhibition, DMSO for 0% inhibition).
- Incubation and Readout: Incubate plates for 72 hours in a standard gas mixture (90% N₂, 5% O₂, 5% CO₂) at 37°C. Quantify parasite viability using a DNA-binding dye like SYBR Green I and measure fluorescence.
- Data Analysis: Calculate percentage growth inhibition relative to controls. Fit dose-response curves to determine the half-maximal inhibitory concentration (IC₅₀) for each compound.
Protocol 2: Target Deconvolution via Resistance Generation and Whole-Genome Sequencing (WGS)
- Resistance Selection: Subject the P. falciparum culture to a sub-lethal concentration of the phenotypic hit compound. Gradually increase the drug pressure over several weeks until resistant parasites emerge [12].
- Clonal Isolation: Isplicate single parasite clones from the resistant population via limiting dilution.
- Genomic DNA Preparation: Extract genomic DNA from the resistant clones and the parent (drug-sensitive) clone.
- Whole-Genome Sequencing: Sequence the genomes of resistant and parent clones to high coverage using next-generation sequencing platforms.
- Variant Analysis: Bioinformatically identify single nucleotide polymorphisms (SNPs), insertions, or deletions (Indels) that are unique to or enriched in all resistant clones. The gene harboring the mutation is the most likely molecular target of the compound [12].
Protocol 3: Biochemical Assay for a Validated Target (e.g., an Enzyme)
- Protein Purification: Express and purify the recombinant Plasmodium target protein (e.g., in E. coli or using endogenous methods from parasites [22]).
- Assay Optimization: Develop a robust biochemical assay (e.g., fluorescence-based, radiometric) to measure the enzyme's activity in the presence of its substrate.
- Compound Screening: Incubate the enzyme with test compounds across a concentration range, then initiate the reaction with the substrate.
- Activity Measurement: Monitor the generation of the product or consumption of the substrate over time. Calculate the percentage of enzyme inhibition and determine the IC₅₀ for each compound [12].
The biological roadmap provided by the Plasmodium lifecycle is indispensable for structuring the modern antimalarial drug discovery pipeline. By aligning Target Candidate Profiles (TCPs) with specific parasite vulnerabilities, the research community can systematically develop the diverse therapeutic tools needed for eradication. The debate between phenotypic and target-based screening is not about choosing a single winner; the most productive strategy is a synergistic one [19]. Phenotypic screening excels at identifying novel, cell-active chemical starting points with new mechanisms of action, while target-based approaches provide a rational path for optimizing hits and overcoming resistance, especially as high-resolution structural information becomes available [22]. The future of antimalarial discovery lies in leveraging the strengths of both methodologies, using the clear guidance of the TCP framework to deliver the next generation of medicines that will ultimately help eradicate this devastating disease.
Screening in Action: Methodologies and Antimalarial Case Studies
Phenotypic screening has established itself as a powerful paradigm in antimalarial drug discovery, consistently outperforming target-based approaches in delivering clinical candidates over the past two decades [7]. This methodology involves screening compounds against whole parasite cells, preserving the complex biological context of the pathogen and enabling the identification of novel therapeutic agents without prior knowledge of specific molecular targets [4]. The trajectory of phenotypic screening technologies has evolved significantly from initial focus on the symptomatic asexual blood stage (ABS) to encompass sophisticated multi-stage assays that address the entire Plasmodium lifecycle [5]. This technological progression responds to the urgent need for medicines that not only treat symptomatic infection but also prevent transmission and target dormant liver stages, addressing key challenges in malaria eradication efforts [13] [23].
The growing emphasis on multi-stage activity represents a paradigm shift in antimalarial development. With resistance emerging against front-line artemisinin-based combination therapies (ACTs) and the only licensed transmission-blocking drug, primaquine, presenting safety concerns for glucose-6-phosphate dehydrogenase (G6PD) deficient individuals, the demand for compounds active across multiple parasite stages has intensified [5] [23]. Contemporary phenotypic screening platforms now regularly incorporate liver stage, sexual gametocyte stage, and mosquito stage assays alongside traditional ABS evaluations, providing a comprehensive assessment of compound activity throughout the parasite's complex lifecycle [5].
Technological Progression: From ABS-Centric to Multi-Stage Platforms
Historical Focus: Asexual Blood Stage Screening
Asexual blood stage screening constituted the foundation of phenotypic antimalarial discovery, primarily because ABS Plasmodium falciparum parasites are readily maintained in vitro using cultured erythrocytes [4]. Early ultra-high throughput screens employed detection methods such as nucleic acid intercalating dyes to stain parasitic DNA, with fluorescence signal serving as a proxy for parasite proliferation [4]. These ABS-focused campaigns successfully identified several clinical candidates, including the spiroindolone KAE609, which demonstrated remarkably rapid parasite clearance kinetics with a median half-life of 0.9 hours and represents the first new scaffold class introduced to the antimalarial chemical space in over 20 years [4].
Despite these successes, conventional ABS proliferation assays faced limitations in capturing important pharmacodynamic properties. Innovative approaches subsequently emerged to address these gaps, such as the invasion assay developed by Linares et al., which distinguished between drugs with rapid, artemisinin-like killing kinetics and those with slower onset of action by measuring the ability of parasites to invade erythrocytes and re-establish infections after compound exposure [4]. This medium-throughput flow cytometry-based assay provided valuable insights into compound killing kinetics that traditional proliferation measurements could not capture.
Expansion to Exo-Erythrocytic Stages
The recognition that effective malaria eradication requires drugs targeting all parasite lifecycle stages spurred development of screening technologies for exo-erythrocytic stages. The parasite's liver stage (LS) represents a critical therapeutic target for causal prophylaxis and prevention of relapsing infections caused by P. vivax and P. ovale hypnozoites [4] [5]. Similarly, the sexual gametocyte stages represent key targets for transmission-blocking interventions aimed at interrupting malaria spread [23].
Technological advances have enabled high-throughput screening against these previously challenging stages. For liver stages, robust in vitro hepatocyte infection models now facilitate screening against initial infection and dormant forms [4]. For gametocytes, the development of transgenic parasite lines expressing luciferase viability reporters, such as the NF54/iGP1_RE9Hulg8 line described in a 2025 Nature Communications publication, has revolutionized transmission-blocking drug discovery [23]. This engineered line conditionally produces large numbers of stage V gametocytes and enables quantitative assessment of compound effects through bioluminescence measurement, overcoming historical challenges associated with gametocyte quiescence and low metabolic activity [23].
Table 1: Evolution of Phenotypic Screening Technologies for Different Parasite Stages
| Parasite Stage | Historical Screening Methods | Modern Advanced Platforms | Key Technological Innovations |
|---|---|---|---|
| Asexual Blood Stage (ABS) | Nucleic acid staining (SYBR Green), enzymatic viability assays | Image-based high-content screening, invasion assays | High-resolution fluorescence microscopy, automated image analysis (Columbus) [1] |
| Liver Stage (LS) | Limited in vivo models, low-throughput microscopy | In vitro hepatocyte infection models, luciferase reporters | Humanized liver models, reporter parasite lines [5] |
| Gametocyte Stage | Morphological assessment, mosquito feeding assays | Transgenic luciferase lines (NF54/iGP1_RE9Hulg8), metabolic assays | Conditional gametocyte production, bioluminescence imaging, humanized mouse models [23] |
| Multi-Stage | Sequential single-stage screening | Integrated lifecycle platforms, AI-powered prediction (MalariaFlow) | Unified reporter systems, computational modeling, high-throughput multi-parameter readouts [24] [23] |
Contemporary Multi-Stage Screening Platforms and Workflows
Advanced Gametocyte Screening Platforms
The establishment of robust gametocyte screening platforms represents one of the most significant advances in phenotypic antimalarial discovery. The comprehensive pipeline described by T. G. et al. (2025) integrates in vitro discovery with in vivo testing through several innovative components [23]. This platform utilizes transgenic NF54/iGP1_RE9Hulg8 parasites engineered to conditionally produce large numbers of stage V gametocytes expressing a red-shifted firefly luciferase viability reporter, enabling highly quantitative screening [23]. The incorporation of a humanized mouse model that can be infected with pure stage V gametocytes allows for unprecedented assessment of in vivo gametocyte killing and clearance kinetics using whole animal bioluminescence imaging [23].
This integrated approach addresses critical historical limitations in transmission-blocking drug discovery, including the difficulty of obtaining pure, synchronous stage V gametocytes in sufficient quantities for screening, and the lack of animal models suitable for evaluating transmission-blocking potential in vivo [23]. The platform enables researchers to seamlessly transition from in vitro identification of gametocytocidal compounds to in vivo validation of transmission-blocking efficacy, significantly accelerating the development of much-needed transmission-blocking agents [23].
Diagram 1: Integrated transmission-blocking screening workflow combining in vitro and in vivo components [23].
Artificial Intelligence-Enhanced Phenotypic Screening
Artificial intelligence has revolutionized phenotypic screening by enabling sophisticated analysis of complex datasets and prediction of compound activity across multiple parasite stages. The MalariaFlow platform exemplifies this integration, representing a comprehensive deep learning resource for multi-stage phenotypic antimalarial discovery [24]. This platform systematically compares multiple machine learning and deep learning algorithms—including fingerprint-based models (RF::Morgan, XGBoost:Morgan), graph-based deep learning models (GCN, GAT, MPNN, Attentive FP), and co-representation deep learning models (FP-GNN, HiGNN, FG-BERT)—to predict antimalarial activity across ten Plasmodium phenotypes and three lifecycle stages [24].
Notably, the FP-GNN (Fingerprints and Graph Neural Networks) model demonstrated superior predictive performance, achieving an overall AUROC of 0.900 and accurately capturing key structural features responsible for multi-stage activities [24]. This hybrid approach outperformed both classical fingerprint-based machine learning and pure graph-based deep learning models by effectively integrating domain-specific chemical knowledge with structural information [24]. The platform successfully identified novel triple-stage antimalarial hits that were subsequently validated through experimental testing, demonstrating the practical utility of AI-enhanced approaches in accelerating antimalarial discovery [24].
Comparative Performance: Phenotypic vs. Target-Based Screening
The impact of phenotypic versus target-based screening approaches in antimalarial drug discovery has been quantitatively assessed over the past two decades (2005-2025), with phenotypic screening demonstrating clear advantage in delivering clinical candidates [7]. This performance differential stems from several inherent advantages of phenotypic screening, including natural elimination of non-membrane permeable compounds, ability to identify drugs with cooperative effects or multiple targets, and independence from prior target knowledge [4]. Perhaps most importantly, phenotypic screening significantly increases the probability of identifying first-in-class drugs operating through novel mechanisms of action, a critical consideration in light of emerging resistance to all front-line antimalarials [4].
Table 2: Key Multi-Stage Active Compounds Identified Through Phenotypic Screening
| Compound | Asexual Blood Stage (IC₅₀) | Liver Stage Activity | Gametocyte Stage Activity | Primary Molecular Target |
|---|---|---|---|---|
| MMV030084 | Nanomolar activity [5] | Active against P. berghei [5] | Transmission-blocking activity [5] | PKG [5] |
| WM382 | Nanomolar activity [5] | Reduces hepatic merozoite viability [5] | Inhibits oocyst development [5] | Plasmepsin IX/X [5] |
| SGI-1027 | Low-nanomolar potency [5] | Not specified | Active against early-stage gametocytes [5] | DNA methyltransferase [5] |
| ONX-0914 | IC₅₀ < 500 nM [1] | Not specified | Not specified | Not specified [1] |
Essential Research Reagents and Methodologies
Successful implementation of contemporary phenotypic screening platforms requires specialized reagents and methodologies optimized for specific parasite stages and detection modalities. The following toolkit represents critical components referenced in recent high-impact studies:
Table 3: Essential Research Reagent Solutions for Phenotypic Screening
| Reagent / Material | Function | Application Examples |
|---|---|---|
| Transgenic NF54/iGP1_RE9Hulg8 parasites | Conditionally produce stage V gametocytes with luciferase reporter | In vitro and in vivo gametocytocidal screening [23] |
| Wheat germ agglutinin-Alexa Fluor 488 | Fluorescent staining of erythrocytes | Image-based high-throughput screening [1] |
| Hoechst 33342 | Nucleic acid staining for parasite detection | Quantification of parasite proliferation in ABS assays [1] |
| N-Acetyl-glucosamine (GlcNAc) | Selective elimination of asexual parasites | Production of synchronous gametocyte cultures [23] |
| Humanized NODscidIL2Rγnull mice | In vivo model for human Plasmodium stages | Evaluation of transmission-blocking efficacy [23] |
| CRISPRi/dCas9 systems | Gene knockdown for target validation | Functional genomics and mechanism of action studies [25] |
Detailed Experimental Protocols
Image-Based High-Throughput Screening Protocol
The image-based antimalarial screening protocol optimized by C. K. et al. (2025) provides a robust methodology for quantitative phenotypic assessment [1]. This protocol involves dispensing synchronized Plasmodium falciparum cultures (1% schizont-stage parasites at 2% hematocrit) into 384-well plates containing test compounds, followed by 72-hour incubation under standard malaria culture conditions (37°C, 1% O₂, 5% CO₂ in N₂) [1]. Post-incubation, plates are diluted to 0.02% hematocrit and stained with a solution containing wheat germ agglutinin-Alexa Fluor 488 conjugate (1 µg/mL) for erythrocyte membrane staining and Hoechst 33342 (0.625 µg/mL) in 4% paraformaldehyde for parasite nucleic acid staining and fixation [1]. Image acquisition is performed using high-content imaging systems such as the Operetta CLS with 40× water immersion lens, capturing nine microscopy fields per well with a resolution of 0.299 µm pixel size [1]. Subsequent image analysis using platforms like Columbus software enables automated quantification of parasite viability and growth inhibition based on fluorescence signals [1].
Integrated Transmission-Blocking Assessment Protocol
The comprehensive transmission-blocking assessment protocol combines in vitro and in vivo components as detailed in the 2025 Nature Communications publication [23]. For in vitro screening, pure stage V gametocytes from the NF54/iGP1_RE9Hulg8 line are exposed to test compounds for 48-72 hours, followed by quantification of viability using luciferase activity measurement [23]. For in vivo assessment, humanized NODscidIL2Rγnull mice are infected with pure stage V gametocytes via intravenous injection, followed by treatment with test compounds and whole animal bioluminescence imaging to monitor gametocyte clearance kinetics [23]. Finally, transmission-blocking efficacy is confirmed using the standard membrane feeding assay (SMFA), where Anopheles mosquitoes feed on gametocyte-infected blood from treated mice, with subsequent assessment of oocyst development in mosquito midguts [23]. This multi-faceted approach provides comprehensive validation of transmission-blocking activity across both mammalian and mosquito lifecycle stages.
Diagram 2: Multi-stage phenotypic screening workflow integrating in vitro and in vivo validation [23].
Phenotypic screening technologies for antimalarial discovery have undergone remarkable evolution, transitioning from ABS-centric approaches to comprehensive multi-stage platforms that address the entire Plasmodium lifecycle. This technological progression, coupled with integration of artificial intelligence and sophisticated in vivo models, has significantly enhanced our ability to identify compounds with potential to not only treat symptomatic malaria but also prevent transmission and target dormant liver stages [5] [23]. The demonstrated superiority of phenotypic over target-based approaches in delivering clinical candidates underscores the importance of maintaining complex biological context in early discovery phases [7].
Future developments in phenotypic screening will likely focus on increasing throughput and information content while further integrating computational approaches. Platforms like MalariaFlow that leverage deep learning for multi-stage activity prediction represent the vanguard of this trend, potentially enabling virtual screening of compound libraries against multiple parasite stages before resource-intensive experimental validation [24]. Additionally, continued refinement of humanized mouse models and reporter parasite lines will enhance the translational relevance of preclinical findings, potentially reducing attrition rates in later development stages [23]. As resistance to current therapies continues to spread, these advanced phenotypic screening technologies will play an increasingly vital role in replenishing the antimalarial development pipeline with multi-stage candidates capable of addressing the evolving challenges of malaria control and eradication.
The emergence and spread of Plasmodium falciparum resistance to first-line artemisinin-based combination therapies represents one of the most pressing challenges in malaria control [4] [26]. This resistance threat, coupled with the limited arsenal of drugs targeting multiple parasite life cycle stages, has intensified the search for new antimalarial chemotypes with novel mechanisms of action [4] [27]. In this context, phenotypic high-throughput screening has re-emerged as a powerful strategy for identifying novel antimalarial compounds, contrasting with target-based approaches that require prior knowledge of specific molecular targets [4] [28].
Phenotypic screens offer several distinct advantages for antimalarial discovery: they identify compounds active against the whole parasite, naturally eliminate non-membrane permeable compounds, can reveal drugs with cooperative effects or multiple targets, and most importantly, can identify first-in-class drugs operating through completely new mechanisms of action without requiring target pre-specification [4] [28]. This review examines three success stories from phenotypic screening campaigns—KAE609 (cipargamin), KAF156 (ganaplacide), and MMV030084—that exemplify the power of this approach for delivering promising antimalarial candidates with novel mechanisms of action.
Comparative Analysis of Phenotypic vs. Target-Based Screening
Table 1: Key characteristics of phenotypic versus target-based screening approaches in antimalarial drug discovery.
| Feature | Phenotypic Screening | Target-Based Screening |
|---|---|---|
| Screening paradigm | Whole-cell screening against entire parasites | Screening against purified target proteins or enzymes |
| Target knowledge requirement | No prior target knowledge needed | Requires identified and validated molecular target |
| Membrane permeability | Naturally selects for compounds that can penetrate cells | May identify potent enzyme inhibitors that cannot reach intracellular targets |
| Target identification | Requires subsequent target deconvolution efforts | Target is known from the outset |
| Mechanism complexity | Can identify compounds with multi-target or cooperative effects | Typically focuses on single-target mechanisms |
| Success rate for first-in-class drugs | Historically higher for identifying first-in-class medicines | More successful for follower drugs [28] |
The debate between screening strategies remains highly relevant in antimalarial research. While target-based approaches enable rational drug design and straightforward optimization, phenotypic screens have consistently delivered novel chemotypes with unexpected mechanisms of action [28]. The success of phenotypic screening is particularly valuable for malaria parasites, which have complex life cycles with stage-specific biological processes that may not be adequately targeted by focusing on previously validated targets [4].
Compound Profiles and Comparative Data
KAE609 (Cipargamin)
KAE609 represents a breakthrough in antimalarial therapy as the first spiroindolone class compound to advance to clinical trials [29]. Discovered in a phenotypic screen designed to identify compounds that rapidly clear intracellular P. falciparum from human red blood cells, KAE609 exhibited exceptional potency and a remarkably fast parasite clearance profile [28]. Structure-activity relationship studies demonstrated that the (1R,3S) configuration was essential for antimalarial activity, with fluorine and chlorine substituents on the benzene ring contributing to increased potency and metabolic stability [29].
Table 2: Key experimental data for KAE609 (cipargamin) across multiple parasite stages and models.
| Parameter | Value | Experimental Context |
|---|---|---|
| Asexual blood stage IC₅₀ | 0.5 - 550 pM [30] | P. falciparum strains |
| Parasite clearance half-life | 0.9 hours [4] | Clinical trial data |
| Transmission-blocking concentration | Complete block at 500 nM [26] | Standard membrane feeding assay |
| In vivo efficacy (P. berghei) | 99.6% reduction at 30 mg/kg [29] | Mouse model, single oral dose |
| Human elimination half-life | 24.0 ± 7.6 hours [29] | Phase 1 clinical trial |
| Gametocyte IC₅₀ (male/female) | 115.6/104.9 nM [26] | Artemisinin-resistant isolates |
The molecular target of KAE609 was identified through resistance selection studies as PfATP4, a parasite plasma membrane P-type ATPase ion transporter that maintains sodium homeostasis [29] [30]. Inhibition of PfATP4 disrupts sodium efflux, leading to increased intracellular sodium concentrations, parasite swelling, and eventual death [26] [29]. This mechanism represents a previously unexploited target class in antimalarial therapy [30].
KAF156 (Ganaplacide)
KAF156 belongs to the novel imidazolopiperazine class and was discovered through phenotypic screening against asexual blood stage parasites [31] [32]. This compound demonstrates broad-stage activity with potent action against liver, asexual blood, and sexual transmission stages of the parasite, making it particularly valuable for both treatment and prevention [26] [33]. Its clinical development represents the first compound from the imidazolopiperazine class to advance to phase IIb combination studies [33].
Table 3: Key experimental data for KAF156 (ganaplacide) across multiple parasite stages.
| Parameter | Value | Experimental Context |
|---|---|---|
| Asexual blood stage IC₅₀ | 5.6 - 7.7 nM [26] | Artemisinin-resistant isolates |
| Gametocyte IC₅₀ (male/female) | 6.9/47.5 nM [26] | Artemisinin-resistant isolates |
| Liver stage activity | Active [32] | Causal prophylactic efficacy in CHMI model |
| Transmission-blocking | Active at 500 nM [32] | In vitro and in vivo models |
| Strain coverage | Pan-active against Plasmodium species [29] | Including P. falciparum and P. vivax |
Unlike KAE609, the precise molecular target of KAF156 remains to be fully elucidated, highlighting one of the challenges of phenotypic screening approaches [26]. However, studies with the related imidazolopiperazine compound GNF179 suggest this class may inhibit protein trafficking, block establishment of new permeation pathways, and cause endoplasmic reticulum expansion in parasites [26] [29]. The unknown target underscores how phenotypic screens can identify valuable chemotypes without requiring immediate mechanism understanding [28].
MMV030084
MMV030084 is a trisubstituted imidazole compound identified from phenotypic screening efforts that exhibits a remarkable multi-stage activity profile [34]. It potently inhibits hepatocyte invasion by Plasmodium sporozoites, merozoite egress from asexual blood stage schizonts, and male gamete exflagellation, providing prophylactic, blood stage, and transmission-blocking antiplasmodial activity [34].
Table 4: Key characteristics and experimental findings for MMV030084.
| Parameter | Details | Significance |
|---|---|---|
| Chemical class | Trisubstituted imidazole [34] | Novel chemotype |
| Primary target | cGMP-dependent protein kinase (PKG) [34] | Validated through multiple orthogonal approaches |
| Resistance profile | PKG itself never mutated under drug pressure [34] | Suggests potential resistance-refractory characteristics |
| Stage specificity | Prophylactic, blood stage, and transmission-blocking [34] | Multi-stage activity |
| Secondary resistance mediator | Tyrosine kinase-like protein 3 (TKL3) [34] | Low-level resistance mediator |
Comprehensive target deconvolution efforts for MMV030084 employed metabolomic, phosphoproteomic, and chemoproteomic studies, validated with conditional knockdown parasites, molecular docking, and recombinant kinase assays [34]. These orthogonal approaches identified cGMP-dependent protein kinase (PKG) as the primary target, which aligns perfectly with the compound's multi-stage activity profile since PKG is known to play essential roles in Plasmodium invasion and egress from host cells [34].
Experimental Protocols and Methodologies
Phenotypic Screening Protocols
The discovery of KAE609 and KAF156 stemmed from robust phenotypic screening platforms against asexual blood stage parasites [4] [28]. The standard approach utilizes P. falciparum cultures in human erythrocytes with detection methods including:
- SYBR Green I-based fluorescence assays that measure parasite nucleic acid content [26] [27]
- Luciferase reporter systems in genetically modified parasite lines [4] [27]
- Enzymatic methods linking parasite viability to enzyme activity [4]
These assays are performed in microtiter plates (96- to 1536-well formats) with typical compound incubation periods of 72 hours for initial screening [4] [27]. For liver stage screening, the generation of P. berghei expressing a GFP-Luc reporter (Pb-Luc) enabled development of sensitive, high-throughput luciferase-based assays [27]. This technical advancement was crucial for identifying chemoprotective compounds like KAF156 that target early exoerythrocytic forms [27] [28].
Target Deconvolution Methods
Once active compounds are identified through phenotypic screening, substantial effort is required to determine their mechanisms of action:
- In vitro evolution and whole-genome sequencing: Parasites or yeast models are subjected to drug pressure, with subsequent genomic sequencing of resistant clones to identify mutations conferring resistance [30]
- Metabolomic and phosphoproteomic profiling: Global analysis of metabolic or phosphorylation changes in response to compound treatment [34]
- Chemoproteomic approaches: Direct assessment of compound-protein interactions [34]
- Functional validation: Using tools like CRISPR/Cas to introduce identified mutations and confirm their role in resistance [30]
Diagram 1: Target deconvolution workflow for phenotypic screening hits. The process begins with compound identification and proceeds through multiple parallel approaches for target identification and mechanism elucidation.
The Scientist's Toolkit: Essential Research Reagents
Table 5: Key research reagents and their applications in antimalarial phenotypic screening and validation.
| Reagent/Cell Line | Function/Application | Key Features |
|---|---|---|
| P. falciparum Dd2/3D7 strains | Standard strains for asexual blood stage screening [27] | Drug-sensitive (3D7) and resistant (Dd2) reference lines |
| P. berghei GFP-Luc reporter | Liver stage screening and in vivo validation [27] | Enables high-throughput luciferase-based quantification |
| SYBR Green I dye | Nucleic acid staining for parasite proliferation [27] | Fluorescent detection of parasite growth in high-throughput formats |
| ABC16-Monster yeast strain | Target identification and resistance studies [30] | Lacks 16 ABC transporters, making it more susceptible to compounds |
| pH-sensitive GFP (pHluorin) | Intracellular pH measurement [30] | Detects cytoplasmic pH changes in response to ion transport inhibitors |
| PfDGFA (Dual Gamete Formation Assay) | Transmission-blocking activity assessment [26] | Measures exflagellation (male) and Pfs25 expression (female) |
Mechanisms of Action and Signaling Pathways
The three compounds reviewed here exemplify the diverse mechanisms that can be discovered through phenotypic screening:
KAE609 targets PfATP4, a sodium efflux pump located on the parasite plasma membrane [29] [30]. Inhibition causes disruption of sodium homeostasis, leading to increased intracellular sodium, osmotic swelling, and ultimately parasite death [26]. This mechanism also affects cytosolic pH, further contributing to the lethal effect on parasites [30].
MMV030084 inhibits cGMP-dependent protein kinase (PKG), a key signaling enzyme in malaria parasites [34]. PKG plays essential roles in multiple cellular processes including calcium signaling, which regulates actomyosin motor function during invasion and egress [34]. The central position of PKG in these critical pathways explains the multi-stage activity observed with this compound.
KAF156's mechanism, while not fully elucidated, appears to involve disruption of protein trafficking and endoplasmic reticulum function based on studies with related compounds [26] [29]. The expansion of endoplasmic reticulum and inhibition of establishment of new permeation pathways suggest interference with fundamental cellular processes essential across parasite life cycle stages.
Diagram 2: Diverse mechanisms of action identified through phenotypic screening. Despite different molecular targets, all three compounds exhibit activity across multiple parasite life cycle stages.
The success stories of KAE609, KAF156, and MMV030084 powerfully demonstrate the value of phenotypic screening in antimalarial drug discovery. These compounds, identified through whole-cell screening approaches without prior target bias, have yielded:
- Novel chemotypes with exceptional potency against multidrug-resistant parasites
- First-in-class mechanisms targeting previously unexploited biological pathways
- Multi-stage activity profiles addressing treatment, prophylaxis, and transmission-blocking needs
- Clinical candidates that have advanced to address urgent medical needs
While phenotypic screening presents challenges in target deconvolution, the integration of advanced chemical biology approaches—including chemoproteomics, resistance selection with whole-genome sequencing, and functional validation—has created a powerful pipeline for transforming phenotypic hits into well-characterized clinical candidates [34] [30] [28].
The continued evolution of screening technologies, including improved liver stage and transmission-blocking assays, coupled with the public availability of large-scale screening data, promises to further accelerate the discovery of next-generation antimalarials [27]. As resistance to current therapies continues to spread, the innovative application of phenotypic screening will remain essential for replenishing the antimalarial pipeline and achieving malaria eradication goals.
In the relentless battle against malaria, the emergence of resistance to front-line artemisinin-based combination therapies (ACTs) underscores an urgent need for novel antimalarials with new mechanisms of action [7] [5]. Antimalarial drug discovery has historically been dominated by phenotypic screening, an approach that has successfully delivered clinical candidates without requiring prior knowledge of the drug's molecular target [35]. However, with advances in technology, target-based strategies that leverage genomic and structural data are experiencing a renaissance, offering new avenues for developing selective and potent therapeutics against Plasmodium falciparum, the deadliest malaria-causing parasite [35].
This guide objectively compares the performance of target-based approaches against alternative strategies by examining foundational methodologies, key experimental data, and the essential toolkit that empowers modern antimalarial research.
Genomic Foundations for Target Identification and Validation
The completion of the P. falciparum genome sequence in 2002 promised a wealth of potential new drug targets [35]. Genomic approaches now enable the systematic identification and validation of essential genes and pathways, providing a scientific basis for target-based discovery.
Functional Genomic Screening for Drug-Resistance Genes
A powerful functional screening approach identifies drug-resistance genes by directly interrogating parasite genomics [36]. This method involves creating a high-coverage genomic library from a drug-resistant strain and introducing it into a drug-sensitive strain. Subsequent drug screening isolates resistant parasites, allowing for the identification of the responsible gene from the transferred DNA [36].
Experimental Protocol:
- Library Construction: Genomic DNA from a donor strain (e.g., Dd2 for chloroquine resistance) is partially digested, and large fragments (>10 kb) are cloned into a centromere plasmid vector (e.g., pFCENv1) to ensure stable, single-copy maintenance in parasites [36].
- Transfection: The plasmid library is transfected into synchronized schizonts of a sensitive recipient strain (e.g., 3D7), and transgenic parasites are selected with a marker drug like pyrimethamine [36].
- Drug Selection & Cloning: The library of transfected parasites is subjected to the antimalarial drug of interest (e.g., 20 nM chloroquine). Surviving parasites are cloned, and their IC50 values are determined to confirm resistance [36].
- Target Identification: The plasmid is recovered from resistant clones, and the inserted DNA fragment is sequenced to identify the gene conferring resistance [36].
Table 1: Key Metrics from a Functional Genomic Screening Pilot Study [36]
| Metric | Result | Implication |
|---|---|---|
| Transfection Efficiency | ~500 independent clones per 5 µg DNA | Enables sufficient genomic coverage |
| Average Insert Size | 25.9 kb | Contains ~6 genes per fragment |
| Genomic Coverage | ~2.6 genome equivalents from 3 libraries | High probability of identifying true resistance genes |
| Identified Gene (Chloroquine) | pfcrt (known resistance gene) | Validates method's effectiveness |
| Identified Gene (Mefloquine) | pfmdr7 (novel candidate) | Demonstrates discovery potential |
In Vitro Evolution and Whole-Genome Sequencing
This "reverse chemical genetics" approach is pivotal for validating targets of compounds identified in phenotypic screens [35]. Parasites are exposed to increasing sub-lethal concentrations of a bioactive compound over months until resistance emerges. Subsequent whole-genome sequencing of resistant clones is used to pinpoint specific mutations that reveal the compound's molecular target [35].
Structural Biology in Target-Based Drug Discovery
Structural biology provides the atomic-level resolution needed to design inhibitors with high potency and selectivity, a critical advantage when targeting conserved eukaryotic proteins shared by parasite and host.
Structural Comparison for Selective Inhibitor Design
A comparative analysis of the 20S proteasome's β5 subunit from Plasmodium and humans reveals distinct structural differences that can be exploited for selective drug design [37].
Table 2: Comparative Structural Analysis of Human and Plasmodium Proteasome β5 Subunits [37]
| Feature | Human β5 Subunit | Plasmodium β5 Subunit |
|---|---|---|
| Secondary Structure | Rich in β-sheets, more compact conformation | Higher prevalence of loops, more open and flexible |
| Binding Pocket | Restricted, accommodates only small compounds | Open and flexible, can bind a larger, more diverse array of compounds |
| Theoretical pI | 6.91 (slightly acidic) | 5.40 (more acidic) |
| Instability Index | 43.78 | 51.82 (suggests higher susceptibility to degradation) |
| Ramachandran Favored | 98.5% of residues | 83.3% of residues |
Experimental Protocol: Structural Comparison
- Structure Retrieval: Obtain 3D structures of human and malarial proteasomes from the Protein Data Bank (e.g., PDB ID 7LXV and 7LXT) [37].
- Model Preparation: Energy minimization and loop refinement are performed using software like UCSF Chimera and Discovery Studio [37].
- Structural & Dynamical Analysis: Use online servers like VADAR to analyze secondary structure percentages. Conduct molecular dynamics simulations (e.g., 500 ns) to compare stability and flexibility. Perform Principal Component Analysis (PCA) to understand dominant motion patterns [37].
- Binding Pocket Examination: Visualize and analyze binding pocket residues in molecular visualization tools to understand differences in topology and physicochemical properties [37].
In Situ Structural Biology
Cryo-electron tomography (cryo-ET) bridges the gap between in vitro structural data and physiological context. It allows for the visualization of drug-induced phenotypic changes at molecular resolution directly within the parasite [38].
Experimental Protocol: In Situ Cryo-ET
- Sample Preparation: P. falciparum-infected human erythrocytes are treated with a translation inhibitor (e.g., cabamiquine) or a DMSO vehicle control. Cells are cryo-preserved at various time points [38].
- Lamella Preparation: Cryo-focused ion beam (cryo-FIB) milling is used to create thin (150-200 nm) cellular sections (lamellae) suitable for tomography [38].
- Data Collection & Processing: Tomograms are collected from the lamellae. Ribosome particles are identified and extracted for subtomogram averaging and 3D refinement to achieve high-resolution structures of different ribosomal states [38].
- Analysis: The distribution and conformational states of ribosomes in drug-treated versus untreated parasites are compared to elucidate the compound's molecular mechanism and phenotypic impact within the native cellular environment [38].
Visualization of Key Workflows and Pathways
The following diagrams illustrate core experimental workflows and target mechanisms described in the research.
Functional Genomic Screening Workflow
Structural Comparative Analysis for Selective Inhibition
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of target-based strategies relies on a suite of specialized reagents and computational tools.
Table 3: Key Research Reagent Solutions for Target-Based Antimalarial Discovery
| Reagent / Tool | Function / Application | Example / Specification |
|---|---|---|
| Centromere Plasmid Vectors | Stable maintenance of large DNA inserts in parasites for functional genomic screens [36]. | pFCENv1 vector |
| Cryo-ET Infrastructure | High-resolution structural biology within the native cellular environment [38]. | Cryo-FIB-SEM instrumentation |
| Molecular Dynamics Software | Simulating protein dynamics and stability to compare parasite and human targets [37]. | GROMACS with CHARMM36 force field |
| Integrated Drug Discovery Platforms | Collaborative data management, analysis, and computational screening (e.g., docking) [9]. | CDD Vault with integrated AI tools |
| Protein Data Bank (PDB) | Repository for 3D structural data of proteins and complexes for comparative analysis [39] [37]. | PDB IDs 7LXT, 7LXV, 2WE6 |
| Synchronized Parasite Cultures | Essential for stage-specific assays, transfections, and life-cycle studies [36]. | Highly synchronized schizonts |
Target-based strategies, empowered by rich genomic and structural data, provide a rational and powerful framework for antimalarial drug discovery. The ability to precisely identify and validate targets through functional genomics, and then to design selective inhibitors using high-resolution structural comparisons, offers a complementary and often more direct path to drug development compared to traditional phenotypic screening. While phenotypic approaches have historically been highly productive, the integration of these advanced target-based methods is critical for addressing the persistent challenge of antimalarial resistance and for developing the next generation of irresistible medicines.
Malaria remains one of the world's most devastating infectious diseases, with an estimated 263 million cases and over 600,000 deaths annually, primarily affecting children in endemic regions [1]. The emergence of resistance to artemisinin-based combination therapies (ACTs), the current first-line treatment, underscores the urgent need for new antimalarials with novel mechanisms of action [7] [5]. In response, drug discovery paradigms have evolved from traditional phenotypic and target-based approaches toward a more integrated strategy.
Phenotypic screening involves testing compounds on whole cells or organisms and observing biological effects without prior knowledge of specific molecular targets [8]. This approach has historically outperformed target-based methods for discovering first-in-class medicines, particularly for complex diseases like malaria [21] [20]. Conversely, target-based screening focuses on identifying compounds that interact with a specific, known molecular target believed to be critical to the disease process [8]. Each method possesses distinct strengths and limitations in the context of antimalarial development.
This guide examines the emerging paradigm of target-informed phenotypic screening, which integrates the complementary strengths of both approaches. We objectively compare the performance of traditional methods and the hybrid strategy, supported by experimental data and detailed methodologies from recent antimalarial drug discovery campaigns.
Comparative Analysis of Screening Approaches
Table 1: Performance comparison of antimalarial drug discovery approaches
| Parameter | Phenotypic Screening | Target-Based Screening | Target-Informed Phenotypic Screening |
|---|---|---|---|
| Success Rate for First-in-Class Drugs | Higher; responsible for majority of new antimalarial chemotypes [21] [5] | Lower; but successful for optimizing known drug classes [7] | Emerging as promising for novel chemotypes with known targets [24] |
| Target Identification | Required subsequent deconvolution (challenging) [20] | Defined before screening [8] | Preliminary target hypotheses guide optimized assays |
| Throughput | High for asexual blood stages; variable for other stages [4] [40] | Very high with purified targets [8] | High, with focused libraries |
| Complex Biological Context | Preserved (whole parasite) [4] | Lost (isolated proteins/pathways) [8] | Preserved with target-relevant readouts |
| Hit-to-Lead Optimization | Can be challenging without MOA [20] | Streamlined with structural knowledge [8] | Facilitated by initial target hypotheses |
| Multi-Stage Activity Assessment | Possible with stage-specific assays [40] [5] | Limited to single target relevance | Designed for multi-stage profiling |
| Major Antimalarial Examples | Artemisinin, KAE609, KAF156 [8] [40] | HIV protease inhibitors [8] | WM382 (PMIX/X inhibitor) [5] |
Table 2: Recent multi-stage antimalarial hits discovered via phenotypic screening (2020-2023)
| Compound | Asexual Blood Stage IC₅₀ | Liver Stage Activity | Transmission-Blocking Activity | Identified Molecular Target |
|---|---|---|---|---|
| MMV030084 | Nanomolar range [5] | Active (P. berghei) [5] | Yes (prevents gametocyte activation) [5] | PfPKG [5] |
| WM382 | Nanomolar range [5] | Delayed hepatic merozoite egress [5] | Inhibits oocyst development [5] | Plasmepsin IX/X [5] |
| SGI-1027 | Low nanomolar range [5] | Not specified | Active against early-stage gametocytes [5] | DNA methyltransferase [5] |
The Integrated Workflow: Target-Informed Phenotypic Screening
Target-informed phenotypic screening represents a hybrid approach that leverages prior knowledge of potential molecular targets to design more intelligent phenotypic assays and interpret screening outcomes. This methodology maintains the biological complexity of phenotypic screening while incorporating the mechanistic insights typically associated with target-based approaches.
Diagram 1: Target-informed phenotypic screening workflow. This integrated approach begins with target hypothesis generation and proceeds through focused library design to multi-stage phenotypic screening, culminating in confirmed actives with known targets.
The fundamental principle of this approach is the preservation of biological complexity while incorporating target-focused intelligence at critical decision points. As illustrated in Diagram 1, the process begins with target hypothesis generation through genomic analysis, essential pathway identification, and resistance mutation mapping [24] [5]. This preliminary target intelligence informs the design of focused compound libraries and assay conditions that maximize the likelihood of identifying compounds with desired mechanisms of action.
During phenotypic screening, this target awareness enables researchers to incorporate specific biomarkers and secondary assays that provide early mechanistic insights while maintaining the unbiased nature of phenotypic discovery [40] [20]. For example, screening libraries against parasites expressing fluorescently tagged versions of potential target proteins can simultaneously assess compound efficacy and begin target engagement analysis.
Experimental Protocols and Methodologies
Phenotypic High-Throughput Screening for Asexual Blood Stages
Objective: To identify compounds with inhibitory activity against Plasmodium falciparum asexual blood stages through image-based screening [4] [1].
Protocol:
- Parasite Culture: Maintain in vitro cultures of P. falciparum strains (e.g., 3D7, NF54 for drug-sensitive; K1, Dd2 for drug-resistant) in human O+ erythrocytes at 2% hematocrit in complete RPMI 1640 medium supplemented with 0.5% Albumax I [1].
- Synchronization: Synchronize parasites at the ring stage using 5% sorbitol treatment to ensure stage-specific compound evaluation [1].
- Compound Treatment:
- Prepare compounds in 384-well plates at a final test concentration of 10 µM (primary screen) or serial dilutions (dose-response).
- Incubate with parasite cultures (1% parasitemia, 2% hematocrit) for 72 hours at 37°C in a mixed gas chamber (1% O₂, 5% CO₂, balance N₂) [1].
- Staining and Imaging:
- Fix and stain with a solution containing 1 µg/mL wheat germ agglutinin-Alexa Fluor 488 (erythrocyte membrane stain) and 0.625 µg/mL Hoechst 33342 (parasite DNA stain) in 4% paraformaldehyde for 20 minutes at room temperature [1].
- Acquire images using high-content imaging systems (e.g., Operetta CLS) with a 40× water immersion lens, capturing 9 fields per well [1].
- Image Analysis: Use automated analysis software (e.g., Columbus) to quantify parasite viability based on nuclear staining intensity and morphological features [1].
Liver Stage Phenotypic Screening
Objective: To identify compounds active against liver-stage parasites, including hypnozoites of P. vivax, for radical cure and prophylactic applications [4] [40].
Protocol:
- Cell Culture: Seed HepG2 or similar hepatoma cells in 384-well plates and culture to 80-90% confluence [40].
- Sporozoite Isolation: Isolate sporozoites from infected Anopheles mosquito salivary glands through manual dissection and mechanical rupture [40].
- Infection and Compound Treatment:
- Infect hepatocyte monolayers with sporozoites (e.g., P. berghei, P. cynomolgi for hypnozoites).
- Add test compounds immediately after infection for prophylactic activity assessment or at later time points for radical cure activity [40].
- Incubation: Maintain infected, compound-treated hepatocytes for 5-7 days with medium changes as needed [40].
- Detection:
- For P. berghei, use luciferase-expressing parasites and measure bioluminescence as a readout for liver stage development [40].
- For P. cynomolgi hypnozoite formation, use immunofluorescence staining with species-specific antibodies and high-content imaging to distinguish hypnozoites from developing liver schizonts [40].
Computational Target Prediction and Validation
Objective: To identify potential molecular targets for hits derived from phenotypic screens using computational approaches [24] [41].
Protocol:
- Molecular Docking:
- Retrieve protein structures from Protein Data Bank (e.g., PDB ID 7TBD for plasmodial protease) [41].
- Prepare protein structures by adding hydrogens, assigning bond orders, and optimizing side chains using tools like UCSF Chimera [41].
- Screen compound libraries (e.g., CMNPD, ZINC) against target binding sites using docking software such as PyRx [41].
- Molecular Dynamics Simulations:
- Perform simulations using AMBER20 software with the General Amber Force Field (GAFF) [41].
- Solvate protein-ligand complexes in a water box and add ions to simulate physiological conditions.
- Run simulations for 100-200 ns to assess complex stability and binding free energies using MM-PBSA/GBSA methods [41].
- Machine Learning Prediction:
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key research reagents for target-informed phenotypic screening in antimalarial discovery
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Parasite Strains | 3D7 (CQ-sensitive), K1 (CQ-resistant), Dd2 (multidrug-resistant), CamWT-C580Y (ART-resistant) [1] | Assessing strain-specific activity and resistance profiling |
| Cell Culture Supplements | Albumax I, Hypoxanthine, Gentamicin [1] | Supporting in vitro parasite growth in human erythrocytes |
| Fluorescent Probes | Hoechst 33342 (DNA stain), Wheat germ agglutinin-Alexa Fluor 488 (erythrocyte membrane) [1] | Enabling high-content imaging and automated quantification |
| Target-Specific Reporters | Luciferase-expressing parasites (e.g., PbGFP-Luccon) [40] | Monitoring liver stage development and compound efficacy |
| Computational Tools | MalariaFlow, Molecular docking software (PyRx), MD simulation software (AMBER20) [24] [41] | Target prediction, virtual screening, and binding validation |
| Compound Libraries | MMV Pandemic Response Box, In-house specialized libraries [1] [5] | Sources of diverse chemical matter for screening campaigns |
Case Studies: Successful Application of Integrated Approaches
WM382: Dual Plasmepsin Inhibitor
The discovery and development of WM382 exemplifies the power of target-informed phenotypic screening. This compound emerged from a focused library of aspartic protease inhibitors screened against P. falciparum blood stages [5]. While the initial screening was phenotypic, the target-focused design of the library enabled rapid identification of plasmepsins IX and X as the molecular targets through cellular thermal shift assays [5].
WM382 demonstrates nanomolar activity against asexual blood stages, delays hepatic merozoite egress, and inhibits oocyst development in mosquitoes, representing true multi-stage antimalarial activity [5]. The precise target knowledge facilitated mechanistic studies revealing that PMX catalyzes activation of proteins in the egress cascade, while PMIX is essential for merozoite invasion [5]. This detailed mechanistic understanding would have been significantly delayed through conventional phenotypic screening alone.
MalariaFlow: Computational Prediction Platform
MalariaFlow represents the computational manifestation of target-informed phenotypic screening. This deep learning platform integrates over 410,000 antimalarial activity data points across ten Plasmodium phenotypes and three life cycle stages [24]. The FP-GNN model achieved the best predictive performance (AUROC=0.900) by fusing molecular fingerprint knowledge with graph neural networks [24].
The platform successfully identified novel triple-stage antimalarial hits that were experimentally validated, demonstrating how computational approaches can prioritize compounds for phenotypic screening [24]. This effectively reverses the traditional screening paradigm by using target-informed predictions to focus experimental resources on the most promising chemical matter.
Diagram 2: Evolution from traditional to integrated screening approaches. Target-informed phenotypic screening merges strengths of both methods, yielding clinical candidates with known mechanisms and multi-stage efficacy.
The integration of target-based intelligence with phenotypic screening represents a maturing of antimalarial drug discovery. This hybrid approach leverages the unbiased nature of phenotypic screening while incorporating mechanistic insights to accelerate lead optimization and candidate selection. Evidence from recent campaigns demonstrates that target-informed phenotypic screening can deliver high-quality chemical matter with defined molecular targets and multi-stage activity [5].
The continued evolution of this paradigm will likely involve even tighter integration of computational and experimental approaches. Machine learning platforms like MalariaFlow [24] and advanced target validation techniques will enable more sophisticated hypothesis generation prior to phenotypic screening. Furthermore, the development of robust assays across all parasite life cycle stages [4] [40] will facilitate the identification of compounds that not only treat symptomatic blood stage infection but also prevent transmission and provide radical cure for relapsing malaria.
As resistance to current therapies continues to emerge, these integrated approaches offer the best promise for delivering the next generation of antimalarial medicines needed to achieve malaria eradication goals.
Navigating Challenges and Optimizing Screening Outcomes
The Target Deconvolution Problem in Phenotypic Screening
Phenotypic screening has re-emerged as a powerful strategy for discovering first-in-class drugs, particularly in antimalarial research where it has identified promising candidates like the spiroindolone KAE609 [4]. However, a significant challenge follows the identification of active compounds: target deconvolution, the process of identifying the specific molecular target(s) through which a hit compound exerts its biological effect [42]. This guide objectively compares the primary experimental strategies employed to solve this problem, providing researchers with a clear framework for selecting the appropriate methodology.
Key Research Reagent Solutions for Target Deconvolution
The following table details essential reagents and materials used in modern target deconvolution workflows.
| Research Reagent / Solution | Primary Function in Target Deconvolution |
|---|---|
| Affinity Beads (e.g., magnetic beads) | Solid support for immobilizing a compound of interest (the "bait") to isolate bound target proteins from a complex biological lysate [43]. |
| Chemical Probes (with alkyne/azide tags) | Minimally modified versions of the hit compound, often incorporating a "click chemistry" handle (e.g., alkyne) for subsequent attachment of a reporter or affinity tag after cellular binding [43]. |
| Photo-reactive Groups (e.g., benzophenone, diazirine) | Moieties incorporated into a probe that form a covalent bond with a target protein upon UV light exposure, crucial for capturing weak or transient interactions [42] [43]. |
| Activity-Based Probes (ABPs) | Bifunctional molecules containing a reactive electrophile that covalently binds to active-site nucleophiles of specific enzyme classes, and a tag for isolation [43]. |
| Mass Spectrometry | Core analytical technology for the unbiased identification and sequencing of proteins isolated via affinity purification, photoaffinity labeling, or stability-based profiling [42] [43]. |
| Cell Painting Assays | A high-content, multiplexed fluorescent imaging technique that stains multiple cellular compartments, generating rich morphological profiles used for phenotypic screening and MoA prediction [44]. |
Comparative Analysis of Target Deconvolution Technologies
No single deconvolution strategy is universally superior; each offers distinct advantages and faces specific limitations. The choice of method depends on factors such as the nature of the compound, the suspected target class, and available resources.
The table below summarizes the core principles, applications, and key requirements of the major deconvolution platforms.
| Technology | Core Principle | Key Applications | Throughput & Scalability | Key Requirement |
|---|---|---|---|---|
| Affinity Chromatography [42] [43] | Immobilized compound acts as "bait" to purify target proteins from a lysate. | Broad-range target identification; "workhorse" technology. | Medium to High | A high-affinity chemical probe that can be immobilized without losing activity. |
| Photoaffinity Labeling (PAL) [42] [43] | A photoreactive probe cross-links to its target upon UV irradiation, capturing transient interactions. | Studying integral membrane proteins; identifying weak/transient binders. | Medium | A trifunctional probe (compound, photoreactive group, enrichment handle). |
| Activity-Based Protein Profiling (ABPP) [43] | Reactive probes covalently label active sites of specific enzyme families (e.g., proteases, hydrolases). | Target discovery when a specific enzyme class is implicated; lead optimization. | Medium to High (for specific enzyme classes) | An electrophilic compound or a promiscuous probe for the enzyme family of interest. |
| Label-Free Profiling (e.g., Thermal Proteome Profiling) [42] | Measures ligand-induced changes in protein thermal stability across the proteome. | Target and off-target identification under native conditions; no probe modification needed. | High (proteome-wide) | Sensitive mass spectrometry; challenges with low-abundance or membrane proteins. |
| Omics & In Vitro Evolution [4] [45] | Genome-wide sequencing of compound-resistant parasites to identify mutations conferring resistance. | Antimalarial research; target identification and resistance mechanism studies. | Medium (requires parasite culture and sequencing) | Generation of resistant parasite lines; can yield background mutations requiring validation. |
| AI-Driven Morphological Profiling [46] [44] | AI/ML analysis of high-content cell images (e.g., Cell Painting) to predict MoA from phenotypic "fingerprints". | Early MoA prediction and triage; virtual screening; toxicity prediction. | Very High (computational) | A large, high-quality image dataset for training models. |
Detailed Experimental Protocols
Affinity Chromatography and Chemical Proteomics
This is a foundational method for isolating direct binding partners [42] [43].
- Step 1: Probe Design and Synthesis. The hit compound is chemically modified to include a linker and an affinity tag (e.g., biotin). To minimize activity loss, a small "clickable" tag like an alkyne can be used, with the bulky biotin added later via click chemistry.
- Step 2: Preparation of Affinity Matrix. The designed probe is immobilized onto a solid support, such as agarose or magnetic beads.
- Step 3: Pull-Down Experiment. The bead-bound probe is incubated with a cellular lysate (e.g., from P. falciparum-infected red blood cells). Unbound proteins are removed through extensive washing.
- Step 4: Target Elution and Identification. Bound proteins are eluted, typically using a competitive amount of the free, unmodified compound or by SDS-PAGE. The eluted proteins are digested into peptides and identified using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS).
In Vitro Evolution and Whole-Genome Scans
A powerful genetic approach widely used in antimalarial research to identify targets and resistance mechanisms [4] [45].
- Step 1: Selection of Resistant Parasites. Plasmodium falciparum cultures (e.g., drug-sensitive strain 3D7) are subjected to sub-lethal doses of the hit compound over multiple generations. Surviving parasites are collected and the process is repeated to select for stable resistant lines.
- Step 2: Genome Sequencing. Genomic DNA is extracted from the resistant parasite population, as well as from the original sensitive parent strain. Whole-genome sequencing (e.g., Illumina sequencing) is performed on both.
- Step 3: Variant Analysis. The sequenced genomes are compared bioinformatically to identify single nucleotide polymorphisms (SNPs), insertions, or deletions that are unique to the resistant population.
- Step 4: Target Validation. The gene harboring the mutation is considered a candidate target. Validation experiments include CRISPR/Cas9 gene editing to reintroduce the mutation into a sensitive strain to confirm it confers resistance, or biochemical assays to confirm direct binding.
AI-Driven Morphological Profiling
This computational approach leverages high-content imaging to predict MoA without physical isolation of targets [46] [44].
- Step 1: High-Content Imaging. Cells (e.g., U2OS or relevant parasitic cells) are treated with the hit compound and stained using a multiplexed assay like Cell Painting, which labels multiple organelles (nucleus, cytoplasm, cytoskeleton, etc.).
- Step 2: Image Feature Extraction. An automated image analysis software (e.g., CellProfiler) or a deep learning model extracts hundreds to thousands of quantitative morphological features from the images, creating a unique "phenotypic fingerprint" for the compound.
- Step 3: Profile Comparison and MoA Inference. The phenotypic fingerprint of the hit compound is compared to a large reference database of profiles from compounds with known MoAs or genetic perturbations. Machine learning models (e.g., Linear Discriminant Analysis) are used to cluster compounds and predict the MoA of the hit based on phenotypic similarity.
- Step 4: Experimental Follow-up. The computational prediction provides a focused hypothesis, which must then be validated using more direct biochemical or genetic methods (e.g., affinity chromatography or CRISPR knockout).
Within the context of antimalarial research, the choice between phenotypic and target-based screening is nuanced. Phenotypic screening has proven highly successful in delivering novel chemotypes like KAE609, which showed a rapid parasite clearance half-life of 0.9 hours [4]. However, the subsequent "target deconvolution problem" presents a significant bottleneck. The described technologies form a modern toolkit to overcome this hurdle.
Affinity-based methods provide direct biochemical evidence of binding, while genetic methods like in vitro evolution have successfully identified targets such as translation elongation factor 2 (eEF2) and phenylalanine tRNA synthetase (PheRS) in Plasmodium [45]. Meanwhile, emerging AI-driven approaches offer a high-throughput way to triage compounds and generate MoA hypotheses early in the discovery pipeline [44]. The integration of these complementary strategies is accelerating the development of novel antimalarials, which is critically needed to combat rising artemisinin resistance and advance the global fight against malaria [4] [14].
Addressing Translation Gaps in Target-Based Discovery
The following table summarizes the core characteristics, strengths, and challenges of phenotypic and target-based screening approaches in antimalarial drug discovery.
Table 1: Comparison of Phenotypic and Target-Based Screening Approaches
| Feature | Phenotypic Screening | Target-Based Screening |
|---|---|---|
| Definition | Identifies compounds that induce a desired effect in whole cells or organisms [8] | Identifies compounds that interact with a specific, purified molecular target [8] |
| Key Advantage | Does not require prior knowledge of the drug target; captures biological complexity [8] [4] | Allows for precise optimization of drug properties; highly efficient and cost-effective [8] |
| Primary Challenge | Can be resource-intensive; mechanism of action may remain unknown [8] | Requires deep understanding of disease biology; risk of failure if target is poorly validated [8] |
| Success in Delivering Antimalarial Clinical Candidates (2005-2025) | Has outperformed the target-based approach [7] | Has delivered fewer clinical candidates in the last two decades [7] |
| Notable Antimalarial Discoveries | Artemisinin, KAE609 [8] [4] | Research ongoing to improve success rates [7] |
Performance Data: Quantitative Comparison and Context
Empirical Success and Clinical Pipeline Output
Over the last two decades (2005-2025), phenotypic screening has outperformed target-based approaches in delivering clinical candidates for antimalarial drugs [7]. This performance gap is particularly critical given the urgent need for new therapies. Current surveillance from Uganda (2019-2024) confirms a troubling decrease in susceptibility of Plasmodium falciparum to key drugs like dihydroartemisinin (DHA) and lumefantrine, underscoring the threat to current first-line artemisinin-based combination therapies (ACTs) [47]. This reality heightens the demand for efficient and productive discovery paradigms.
Experimental Evidence from High-Throughput Screening
A 2025 meta-analysis of High-Throughput Screening (HTS) provides quantitative data on the hit identification efficiency of a phenotypic screening campaign [1]. The study screened an in-house library of 9,547 small molecules against the asexual blood stage of P. falciparum.
Table 2: Hit Identification from a Phenotypic HTS Campaign [1]
| Screening Stage | Number of Compounds | Key Metric |
|---|---|---|
| Initial Primary Screen | 9,547 | Single concentration (10 µM) |
| Selected for Dose-Response | 256 | Top 3% of actives from primary screen |
| Confirmed Active (IC₅₀ < 1 µM) | 157 | ~61% of dose-response compounds |
| Potent Inhibitors (IC₅₀ < 500 nM) | 3 (identified for in vivo testing) | Effective against drug-resistant strains |
This workflow demonstrates the ability of phenotypic HTS to efficiently distill thousands of compounds down to a manageable number of high-quality, potent leads for further development, even against resistant parasite strains [1].
Experimental Protocols: Core Methodologies in Practice
Detailed Protocol: Image-Based Phenotypic Screening
This protocol is adapted from recent HTS and meta-analysis work [1].
- 1. Compound Library Preparation: An in-house library of small molecules is prepared in DMSO and dispensed into 384-well assay plates using automated liquid handlers (e.g., Hummingwell).
- 2. Parasite Culture and Synchronization: Plasmodium falciparum parasites (including drug-sensitive and resistant strains like 3D7, Dd2, and CamWT-C580Y) are cultured in human O+ erythrocytes. Parasites are double-synchronized at the ring stage using 5% sorbitol treatment.
- 3. Drug Sensitivity Assay: Synchronized schizont-stage parasites are dispensed into compound-treated assay plates at 2% hematocrit and 1% parasitemia. The plates are incubated for 72 hours at 37°C in a mixed-gas environment.
- 4. Staining and Image Acquisition: After incubation, the assay plate is diluted and stained with a solution containing:
- Wheat agglutinin–Alexa Fluor 488: Stains the erythrocyte membrane.
- Hoechst 33342: A nucleic acid stain to identify parasitic DNA.
- 4% paraformaldehyde: Fixes the cells. The plate is incubated for 20 minutes at room temperature before imaging.
- 5. High-Content Imaging and Analysis: Nine image fields per well are acquired using a high-content imaging system (e.g., Operetta CLS with a 40x water immersion lens). Automated image analysis software (e.g., Columbus) is used to quantify parasite proliferation and viability based on the fluorescent signals.
Detailed Protocol: Integrated Structural Biology for Target Validation
This protocol illustrates a modern, powerful method to bridge the translation gap for target-based discovery by visualizing drug effects in a native cellular context [38].
- 1. Sample Preparation: Trophozoite-stage P. falciparum-infected human erythrocytes are treated with a compound of interest (e.g., the translation inhibitor cabamiquine) or a DMSO vehicle control.
- 2. Cryo-Preservation and Lamella Preparation: Cells are cryo-preserved. Thin cellular sections (lamellae, 150–200 nm) are prepared using a cryo-focused ion beam scanning electron microscope (cryo-FIB-SEM).
- 3. Cryo-Electron Tomography (Cryo-ET) Data Collection: Tomograms are collected from the lamellae, capturing the native cellular ultrastructure.
- 4. Subtomogram Averaging and 3D Refinement: Native Pf80S ribosome particles are identified and extracted from the tomograms. Multiple rounds of focused 3D classification and refinement are performed to achieve high-resolution structures of different ribosomal states.
- 5. Analysis of Drug-Induced Phenotypic Changes: The distribution and conformational states of ribosomes from drug-treated and untreated parasites are compared. This reveals, at molecular resolution, how the drug perturbs the translation elongation cycle within the native physiological environment of the parasite.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagent Solutions for Featured Antimalarial Screening Experiments
| Research Reagent / Material | Function and Application |
|---|---|
| Synchronized P. falciparum Cultures (3D7, Dd2, etc.) | Provides biologically relevant, stage-specific parasites for phenotypic screening assays [1]. |
| 384-Well Assay Plates | Enables high-throughput, miniaturized screening of compound libraries, reducing reagent costs [1]. |
| Nucleic Acid Stains (e.g., Hoechst 33342, SYBR Green I) | Fluorescent dyes used to label parasitic DNA for viability and proliferation readouts in phenotypic screens [1] [4]. |
| Cryo-FIB-SEM (Focused Ion Beam SEM) | Advanced instrument used to create thin, cryo-preserved lamellae of infected cells for in situ cryo-electron tomography [38]. |
| CDD Vault with Integrated AI Tools | A collaborative platform that democratizes access to predictive modeling, compound management, and data analysis, particularly for researchers in resource-limited settings [9]. |
Visualizing Screening Workflows and Translation Gaps
Phenotypic vs. Target-Based Screening Workflows
Bridging the Gap with Integrated Structural Biology
The empirical data confirms a performance gap, with phenotypic screening contributing more significantly to the antimalarial clinical pipeline over the past 20 years [7]. However, this does not invalidate the target-based approach. Instead, it highlights the critical need for strategies that enhance target validation and bridge the translation gap. Emerging technologies, particularly integrated structural biology methods like in situ cryo-ET, are poised to revolutionize target-based discovery. By enabling the direct visualization of drug effects on native targets within the parasite, these methods can provide the physiological context often missing from traditional biochemical assays, de-risking the development of target-based therapeutics [38]. The future of antimalarial discovery lies not in choosing one approach over the other, but in strategically integrating phenotypic and target-based methods, augmented by powerful new tools, to accelerate the delivery of urgently needed novel medicines.
Hit Validation and the Importance of Chemical Validation for Targets
In antimalarial drug discovery, the high attrition rate of candidate compounds poses a significant challenge to research and development pipelines. Hit validation serves as the critical gateway in this process, rigorously confirming and prioritizing promising candidates from initial screening outputs for further optimization [48]. This process is particularly crucial for eliminating false positives from high-throughput screens and demonstrating that a hit has genuine potential to bind to a target of interest without obvious chemical features that would impede its development into a drug [48].
The validation philosophy differs substantially between the two predominant drug discovery paradigms: phenotypic screening and target-based screening. Phenotypic screens evaluate compound effects on whole cells or entire microorganisms, typically using Plasmodium falciparum-infected red blood cells, without requiring prior knowledge of the specific drug target [4]. In contrast, target-based screens investigate compound effects on purified proteins or defined molecular targets, offering a more mechanism-focused approach from the outset [7] [1]. Understanding how hit validation strategies diverge between these approaches is essential for antimalarial researchers navigating the complex landscape of drug discovery.
Phenotypic Versus Target-Based Antimalarial Screening: A Performance Comparison
Over the past two decades, the antimalarial drug discovery field has witnessed a notable performance disparity between phenotypic and target-based screening approaches. Analyses of clinical candidates revealed during the 2005-2025 period demonstrate that phenotypic-based drug discovery has consistently outperformed the target-based approach in delivering viable antimalarial candidates [7]. This trend persists despite the intuitive appeal of target-based methods, highlighting the complexity of Plasmodium biology and the challenges of predicting compound behavior in whole-parasite systems.
The superior performance of phenotypic screening in antimalarial discovery can be attributed to several inherent advantages. These whole-cell assays automatically eliminate non-membrane permeable compounds from consideration, immediately filtering for molecules capable of penetrating multiple membranes to reach their intracellular targets [4]. Furthermore, phenotypic screens offer the valuable potential to identify compounds with cooperative effects on multiple targets or those operating through entirely novel mechanisms of action—a significant advantage in overcoming existing drug resistance [4]. This approach has yielded notable successes, including the spiroindolone KAE609, which targets P-type cation-transporter ATPase4 (PfATP4) and demonstrates rapid parasiticidal activity with a median parasite clearance half-life of just 0.9 hours in clinical studies [4].
Quantitative Comparison of Screening Approaches
Table 1: Performance Metrics of Phenotypic vs. Target-Based Screening in Antimalarial Discovery (2005-2025)
| Evaluation Metric | Phenotypic Screening | Target-Based Screening |
|---|---|---|
| Clinical candidate output | Higher | Lower [7] |
| Target knowledge requirement | Not required | Essential [4] |
| Membrane permeability assessment | Intrinsic to assay | Requires separate evaluation [4] |
| Novel mechanism identification | High probability | Limited to predefined target [4] |
| Throughput capacity | High for blood stages [4] | Variable |
| Hit-to-lead optimization complexity | Often requires target deconvolution | Straightforward (target known) |
| Resistance to artifacts | Moderate (false positives common) | Higher for well-designed assays |
Target-based approaches, while thus far less productive in terms of clinical output, offer their own strategic advantages. These methods enable more straightforward structure-activity relationship (SAR) development during optimization cycles and provide clear mechanistic understanding from the earliest stages of discovery [7]. To improve the success rate of target-based methods, researchers have proposed several strategies, including better target selection criteria, improved biochemical assay design that more closely mimics physiological conditions, and enhanced chemical library design focused on lead-like compounds with favorable physicochemical properties [7].
Hit Validation Methodologies and Experimental Protocols
The transition from initial screening hits to validated leads requires rigorous experimental assessment across multiple parameters. Both phenotypic and target-based screening approaches employ specialized validation methodologies to confirm genuine pharmacological activity and prioritize compounds for resource-intensive optimization programs.
Phenotypic Hit Validation Workflows
Phenotypic hit validation begins with confirmation of antiparasitic activity in concentration-response assays. Recent optimized protocols utilize image-based screening technologies with double-synchronized Plasmodium falciparum ring-stage parasites (strains 3D7, NF54, K1, Dd2) incubated with compounds for 72 hours in 384-well plates [1]. Following incubation, parasites are stained with a solution containing wheat germ agglutinin–Alexa Fluor 488 conjugate to label erythrocytes and Hoechst 33342 to stain parasitic DNA, then fixed with paraformaldehyde [1]. High-content imaging using systems like Operetta CLS with a 40× water immersion lens captures nine microscopy fields per well, with subsequent image analysis using software such as Columbus to quantify parasite viability and developmental stages [1].
Table 2: Key Research Reagent Solutions for Antimalarial Hit Validation
| Reagent/Resource | Function in Validation | Application Examples |
|---|---|---|
| Wheat germ agglutinin–Alexa Fluor 488 | Erythrocyte membrane staining | Visualizing infected vs. uninfected red blood cells [1] |
| Hoechst 33342 | Nucleic acid intercalating dye | Quantifying parasite burden and developmental stage [1] |
| SYBR Green I | DNA binding dye | Alternative parasite viability assessment [1] |
| CETSA (Cellular Thermal Shift Assay) | Target engagement validation | Confirming drug-target interaction in cellular context [49] |
| Sorbitol synchronization | Parasite stage synchronization | Obtaining homogeneous parasite populations for screening [1] |
| RPMI 1640 with Albumax I | Parasite culture medium | Maintaining Plasmodium falciparum in vitro [1] |
Secondary validation of phenotypic hits includes assessing activity against drug-resistant parasite strains (e.g., Dd2-R539T, CamWT-C580Y) to identify compounds potentially unaffected by existing resistance mechanisms [1]. Additionally, advanced assays can evaluate stage-specific activity and killing kinetics, such as the invasion assay developed by Linares et al., which distinguishes between rapidly parasiticidal compounds (artemisinin-like) and those with slower onset of action by measuring the ability of treated parasites to invade fresh erythrocytes after compound removal [4].
Target-Based Hit Validation Strategies
For target-based approaches, hit validation focuses on confirming direct interaction with the intended molecular target. Techniques such as molecular docking, surface plasmon resonance, and biochemical inhibition assays provide initial confirmation of target engagement [49]. However, the critical advancement in this domain has been the development of cellular target engagement assays, particularly the Cellular Thermal Shift Assay (CETSA).
The CETSA protocol involves treating intact parasites or parasite lysates with compounds of interest, followed by heating to denature and precipitate unbound target proteins [49]. The stabilized target proteins (bound to compounds) remain in solution and are quantified using immunoassays or high-resolution mass spectrometry [49]. For example, a 2024 study applied CETSA to validate engagement of DPP9 in rat tissue, demonstrating dose- and temperature-dependent stabilization ex vivo and in vivo [49]. This methodology provides direct evidence of target engagement within physiologically relevant cellular environments, bridging the gap between biochemical potency and cellular efficacy.
Integrated Validation Frameworks and Decision Support
Modern antimalarial hit validation has evolved toward integrated frameworks that combine multiple data streams to prioritize compounds with the highest probability of success. The combination of high-throughput screening with meta-analysis represents a powerful approach to evidence-based hit selection [50] [1]. This methodology applies systematic filters to identified hits based on novelty, antimalarial activity (IC₅₀), pharmacokinetic properties (Cmax and T1/2), mechanism of action, and safety parameters (CC50, SI, LD50, MTD) [1].
In a recent 2025 study implementing this approach, researchers began with 9,547 compounds subjected to high-throughput screening at 10 µM [1]. From the top 3% (256 compounds), dose-response analysis identified 157 compounds with IC₅₀ values < 1 µM [1]. Subsequent filtering for safety and pharmacokinetic properties narrowed the field to 19 candidates for in vivo assessment in Plasmodium berghei-infected mouse models [1]. This integrated process ultimately identified three potent inhibitors showing >95% suppression of parasitemia, demonstrating the power of combining experimental data with computational prioritization [1].
Visualization of Hit Validation Workflows
The following diagram illustrates the integrated hit validation workflow for antimalarial compound discovery, combining elements from both phenotypic and target-based approaches:
Integrated Hit Validation Workflow in Antimalarial Discovery
Artificial intelligence and machine learning platforms are increasingly incorporated into these validation frameworks, supporting tasks from target prediction and compound prioritization to pharmacokinetic property estimation [49]. For example, recent work demonstrated that integrating pharmacophoric features with protein-ligand interaction data can boost hit enrichment rates by more than 50-fold compared to traditional methods [49]. These computational approaches not only accelerate lead discovery but improve mechanistic interpretability, which has become increasingly important for regulatory confidence and clinical translation [49].
Hit validation represents a critical decision point in antimalarial drug discovery, determining which chemical starting points merit substantial investment in optimization and development. The evidence gathered over the past two decades clearly demonstrates the superior performance of phenotypic screening approaches in delivering clinical candidates for malaria treatment [7]. However, this does not preclude the strategic value of target-based methods, particularly as our understanding of Plasmodium biology advances and technologies for cellular target engagement validation, such as CETSA, become more sophisticated and accessible [49].
The most productive path forward likely involves integrated approaches that leverage the strengths of both paradigms. Phenotypic screening efficiently identifies chemically tractable starting points with whole-cell activity, while target-based validation provides mechanistic insight crucial for rational optimization. Combined with AI-driven prioritization and meta-analysis of compound properties, this integrated framework offers the greatest potential for accelerating the discovery of novel antimalarial agents capable of overcoming existing drug resistance mechanisms [49] [50] [1]. As antimalarial drug discovery continues to evolve, robust hit validation methodologies will remain foundational to converting screening outputs into clinically effective therapeutics that address the persistent global burden of malaria.
A Comparative Analysis of Screening Impact and Future Directions
The relentless evolution of drug-resistant Plasmodium falciparum parasites underscores the critical need for novel antimalarial medicines. Over the past two decades, the antimalarial drug discovery landscape has been dominated by two distinct methodological paradigms: phenotypic screening and target-based screening [7]. Phenotypic screening involves testing compounds against the whole parasite in its cellular context, while target-based screening assays compounds against a specific, purified protein target known to be essential for parasite survival [4]. As resistance to first-line artemisinin-based combination therapies (ACTs) spreads, tracking the tangible output of these approaches—their success in delivering new clinical candidates—is more important than ever for guiding future research investments and strategies [13] [51]. This analysis provides a quantitative comparison of the clinical pipeline impact of phenotypic versus target-based drug discovery approaches for antimalarials from 2005 to the present.
Table 1: Clinical Candidates and Their Discovery Approaches
The following table summarizes key antimalarial drug candidates advanced in the last two decades, categorized by their primary discovery approach.
| Compound / Combination | Discovery Approach | Clinical Status (as of 2025) | Key Attributes / Mechanism | Associated Resistance |
|---|---|---|---|---|
| KAE609 (Cipargamin) [4] | Phenotypic Screening | Clinical Trials | Spiroindolone; targets PfATP4 (rapid parasite clearance) | Novel mechanism, active against resistant strains |
| GanLum (KLU156) [52] | Phenotypic Screening | Phase III (Positive Results) | Ganaplacide (novel mechanism) + Lumefantrine; disrupts parasite protein transport | Effective against mutant parasites resistant to current medicines |
| MMV048 [51] | Phenotypic Screening | Clinical Trials | Novel chemotype; inhibits PfPI4K (blocks parasite development) | Novel mechanism |
| Artemether-Lumefantrine (ACT) [53] | Natural Product Derivation | Marketed (Standard of Care) | Artemisinin derivative + Aryl-aminoalcohol; rapid action + long-acting | Artemisinin partial resistance reported; partner drug resistance threatens efficacy |
| Dihydroartemisinin-Piperaquine (ACT) [53] | Natural Product Derivation | Marketed (Standard of Care) | Artemisinin derivative + Bisquinolone; rapid action + long-acting | Artemisinin partial resistance & Piperaquine resistance reported |
| ST72 [15] | Target-Based (Falcipain-2) | Preclinical | Inhibits falcipain-2 cysteine protease | Effective against Chloroquine-resistant strains |
Experimental Approaches in Action
Phenotypic Screening Protocols
Phenotypic high-throughput screens (HTS) have been a mainstay in antimalarial discovery. A standard protocol involves culturing asexual blood stage P. falciparum parasites in human erythrocytes and exposing them to compound libraries in 384-well or 1536-well formats [4] [14]. After a standard incubation period (typically 72 hours), parasite viability is quantified using methods like DNA-binding fluorescent dyes (e.g., SYBR Green I) or a luciferase reporter gene system [4]. For example, the screen that identified KAE609 used a nucleic acid dye to specifically stain parasite DNA, with hits selected based on a reduction in fluorescence signal, indicating inhibition of parasite growth [4]. More advanced image-based screening protocols can also classify parasites at different developmental stages, providing richer data [14].
Target-Based Screening Protocols
Target-based screening begins with selecting a validated molecular target, such as an enzyme essential for parasite metabolism or development. For instance, the cysteine protease falcipain-2 (FP2) is a key hemoglobinase and an attractive drug target [15]. A detailed protocol for a target-based virtual screen (TBVS) against FP2 involved:
- Target Preparation: Retrieving the 3D crystal structure of FP2 (PDB ID: 3BPF) from the Protein Data Bank and removing water molecules and co-crystallized ligands.
- Compound Library Preparation: Downloading a library of natural compounds (~90,000 molecules) from the ZINC database and filtering them for drug-like properties using tools like SwissADME.
- Molecular Docking: Using software like InstaDock to perform virtual screening, predicting how each compound binds to the active site of FP2 and calculating binding affinity.
- Experimental Validation: Top-ranked virtual hits (e.g., ST72) were then tested in vitro for their ability to inhibit the activity of purified FP2 enzyme, followed by assays to confirm growth inhibition of both chloroquine-sensitive and -resistant P. falciparum strains [15].
Visualizing the Drug Discovery Workflows
The following diagrams illustrate the standard workflows for phenotypic and target-based screening approaches, highlighting their key differences.
Phenotypic Screening Workflow
Target-Based Screening Workflow
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of these screening strategies relies on a suite of specialized reagents and tools.
| Reagent / Tool | Function in Antimalarial Research |
|---|---|
| Synchronized P. falciparum Cultures | Provides a uniform population of parasites at a specific life cycle stage (e.g., rings, trophozoites) for consistent and reproducible screening results. |
| SYBR Green I Dye | A fluorescent nucleic acid stain used in phenotypic HTS to quantify parasite growth and viability by measuring parasite DNA content. |
| Transgenic Luciferase-Expressing Parasites | Engineered parasite lines (e.g., NF54/iGP1_RE9Hulg8) that produce luciferase, enabling highly sensitive, ATP-dependent viability readouts for both asexual and gametocyte stages. |
| Recombinant Target Proteins | Purified malaria proteins (e.g., Falcipain-2, PfPI4K) essential for developing enzymatic assays in target-based screening and validating a compound's mechanism of action. |
| Standard Membrane Feeding Assay (SMFA) | A critical ex vivo assay to evaluate the transmission-blocking potential of drug candidates by assessing their ability to prevent mosquitoes from becoming infected. |
| Computer-Aided Drug Design (CADD) Software | Tools like InstaDock and SwissADME used for in silico (virtual) screening of compound libraries and prediction of drug-like properties. |
Discussion and Clinical Impact Analysis
The data from the clinical pipeline over the last two decades indicates that the phenotypic screening approach has outperformed the target-based approach in delivering clinical candidates [7]. Key successes like KAE609 and the recently reported Phase III success of GanLum underscore the power of whole-cell screens to identify compounds with novel mechanisms and potent activity against multi-drug resistant parasites [52] [4].
A significant advantage of phenotypic screening is its target-agnostic nature, which allows for the discovery of first-in-class drugs acting on novel biological pathways without prior knowledge of the specific molecular target [4]. This is particularly valuable in malaria, where parasite biology is complex and not all essential targets are known or easily druggable. Furthermore, hits from phenotypic screens are inherently cell-permeable and must overcome biological barriers to exert their effect, de-risking one aspect of later development [4].
In contrast, target-based approaches, while rational and enabling highly focused design, have faced challenges. These include difficulties in accurately validating essential drug targets and the failure of target-specific inhibitors to effectively kill the parasite within the cellular context, potentially due to issues with bioavailability or access to the target within the cell [7]. Despite these challenges, target-based strategies, especially when augmented by modern tools like virtual screening as demonstrated with the falcipain-2 inhibitor ST72, continue to provide valuable starting points and can be powerful for lead optimization once a validated target is established [15].
The most promising trend is the strategic integration of both philosophies. For instance, a phenotypic screen can identify a potent compound, which is then followed by target deconvolution to identify its mechanism of action. This knowledge can, in turn, inform a target-based campaign to develop even more potent next-generation inhibitors [51].
Tracking the clinical output of antimalarial drug discovery approaches clearly shows that phenotypic screening has been the more prolific strategy over the past two decades, yielding several novel clinical candidates addressing urgent resistance threats. However, both phenotypic and target-based approaches are mature and valid. The future of antimalarial research lies not in pitting these methods against each other, but in leveraging their complementary strengths. A hybrid model, where phenotypic screening identifies novel chemical starting points and target-based methods facilitate their rational optimization, presents the most robust pathway to replenish the clinical pipeline and combat the persistent challenge of drug-resistant malaria.
The development of novel antimalarial compounds is urgently needed to combat the rise of drug resistance to front-line therapies like artemisinin combination therapies (ACTs). The global health burden of malaria remains devastating, with an estimated 263 million cases and approximately 597,000 deaths annually [1]. Drug discovery efforts to address this crisis have historically relied on two principal screening strategies: phenotypic and target-based approaches [54] [20]. Phenotypic drug discovery (PDD) involves identifying active compounds based on measurable biological responses in whole cells or organisms without prior knowledge of specific molecular targets [54] [20]. In contrast, target-based drug discovery begins with a well-characterized molecular target and uses rational design to develop compounds that modulate its activity [54]. This guide provides a comprehensive, objective comparison of these approaches specifically for antimalarial research, examining their strengths, limitations, and appropriate applications through experimental data and methodological analysis.
Fundamental Principles and Workflows
Phenotypic Screening Workflow
Phenotypic screening for antimalarials evaluates compounds against whole Plasmodium falciparum parasites at various lifecycle stages, primarily focusing on the symptomatic asexual blood stage (ABS). The workflow begins with culturing parasite-infected red blood cells (RBCs) in vitro, followed by compound exposure. Parasite viability is typically measured using nucleic acid staining with dyes like SYBR Green I or Hoechst 33342, followed by high-content imaging or flow cytometry analysis [4] [1]. Recent advancements include image-based screening technologies that can classify parasites at different developmental stages, providing richer data on compound effects [1].
Figure 1: Phenotypic screening workflow for antimalarial discovery. ABS: Asexual Blood Stage.
Target-Based Screening Workflow
Target-based screening begins with selecting a validated molecular target such as an enzyme, receptor, or structural protein essential for parasite survival. The target protein is purified and used to screen compound libraries in biochemical assays measuring binding affinity or functional inhibition [55] [56]. Hits from these screens are then evaluated in cellular systems to confirm activity against whole parasites. This approach relies heavily on prior knowledge of target biology and validation of its essentiality for parasite survival [54].
Figure 2: Target-based screening workflow for antimalarial discovery.
Comprehensive Comparison Table of Screening Approaches
Table 1: Head-to-head comparison of phenotypic vs. target-based screening for antimalarial discovery
| Parameter | Phenotypic Screening | Target-Based Screening |
|---|---|---|
| Primary Screening Objective | Identify compounds inhibiting parasite growth or development [4] | Identify compounds modulating specific molecular target activity [54] |
| Knowledge Requirement | No prior target knowledge needed [4] | Requires validated molecular target [54] |
| Therapeutic Validation | Built-in; compounds must function in cellular context [20] | Separate target validation required [20] |
| Chemical Starting Points | Diverse chemotypes with whole-parasite activity [4] | Target-focused libraries based on binding sites [56] |
| Target Identification | Required after hit identification (target deconvolution) [55] | Defined before screening [54] |
| Throughput Capacity | Ultra-high throughput possible (million+ compounds) [4] [1] | Variable; typically high throughput [1] |
| Technical Complexity | Moderate to high (requires parasite culture) [4] | Variable (biochemical vs. cellular assays) [56] |
| Success Rate (First-in-Class) | Higher for first-in-class antimalarials [20] | Lower for first-in-class; better for follower drugs [20] |
| Major Limitations | Target deconvolution challenging and time-consuming [55] [20] | Requires pre-validated targets; cellular activity not guaranteed [54] |
| Key Antimalarial Examples | Spiroindolone KAE609 (PfATP4 inhibitor) [4] | Dihydroorotate dehydrogenase inhibitors [55] |
Experimental Protocols and Methodologies
Standardized Phenotypic Screening Protocol for Asexual Blood Stages
Principle: This protocol measures compound effects on Plasmodium falciparum asexual blood stage parasites using nucleic acid staining to quantify parasite proliferation inhibition [4] [1].
Materials and Reagents:
- Plasmodium falciparum cultures (sensitive and resistant strains: 3D7, NF54, K1, Dd2, CamWT-C580Y) [1]
- Human O+ erythrocytes and complete RPMI 1640 medium
- Compound libraries dissolved in DMSO
- 384-well assay plates
- Staining solution: 1 µg/mL wheat germ agglutinin-Alexa Fluor 488 conjugate and 0.625 µg/mL Hoechst 33342 in 4% paraformaldehyde [1]
- High-content imaging system (e.g., Operetta CLS with 40× water immersion lens) [1]
Procedure:
- Double-synchronize parasite cultures at ring stage using 5% sorbitol treatment [1].
- Dispense schizont-stage parasites (1% parasitemia, 2% hematocrit) into 384-well plates containing test compounds (10 µM final concentration or dose-response series) [1].
- Incubate plates for 72 hours at 37°C in malaria culture chamber with mixed gas (1% O₂, 5% CO₂ in N₂) [1].
- After incubation, dilute plates to 0.02% hematocrit and stain with staining solution for 20 minutes at room temperature [1].
- Acquire nine microscopy image fields per well using high-content imaging system [1].
- Analyze images using specialized software (e.g., Columbus v2.9) to quantify parasite proliferation based on nucleic acid staining [1].
Data Analysis:
- Calculate percentage inhibition relative to untreated controls
- Determine IC₅₀ values from dose-response curves
- Classify hits based on potency (IC₅₀ < 1 µM) and selectivity [1]
Target Deconvolution Methods for Phenotypic Hits
In Vitro Resistance Generation:
- Principle: Generate resistant parasites by gradual compound exposure; identify resistance-conferring mutations through whole-genome sequencing to implicate molecular target [55].
- Protocol: Expose parasite cultures to sublethal compound concentrations over multiple cycles; isolate resistant clones; sequence genomes and identify nonsynonymous mutations [55].
Chemoproteomic Approaches:
- Principle: Use compound analogs with affinity tags to capture interacting proteins from parasite lysates; identify bound proteins via mass spectrometry [55].
- Protocol: Design and synthesize biotinylated or photoaffinity analogs of active compounds; incubate with parasite lysates; capture on streptavidin beads; analyze bound proteins by LC-MS/MS [55].
Cellular Thermal Shift Assay (CETSA):
- Principle: Measure compound-induced thermal stabilization of target proteins across the proteome; identify stabilized proteins through quantitative proteomics [55].
- Protocol: Treat parasites with compound or DMSO control; heat to different temperatures; lyse cells; quantify soluble proteins by mass spectrometry; identify proteins stabilized by compound treatment [55].
Target-Based Screening Protocol
Principle: This protocol evaluates compound activity against purified malaria target proteins using biochemical assays, followed by validation in cellular systems [56].
Materials and Reagents:
- Purified recombinant target protein (e.g., PfATP4, PfDHODH, PfPKG)
- Compound libraries in DMSO
- Appropriate biochemical assay reagents (substrates, cofactors, detection reagents)
- 384-well assay plates
- Microplate reader or other detection instrument
Procedure:
- Express and purify recombinant target protein from appropriate expression system.
- Develop and optimize biochemical assay measuring target activity (e.g., enzyme inhibition, binding).
- Screen compound library at single concentration (typically 10 µM) or in dose-response.
- Incubate compounds with target and substrates under optimized conditions.
- Measure signal output (fluorescence, luminescence, absorbance) using plate reader.
- Counter-screen against mammalian orthologs to assess selectivity.
- Validate hits in parasite viability assays to confirm cellular activity.
Data Analysis:
- Calculate percentage inhibition relative to controls
- Determine IC₅₀ values from dose-response curves
- Assess selectivity index relative to mammalian orthologs
- Prioritize hits with desired potency, selectivity, and chemical properties
Research Reagent Solutions for Antimalarial Screening
Table 2: Essential research reagents for antimalarial screening approaches
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Parasite Strains | 3D7 (drug-sensitive), K1 (CQ-resistant), Dd2 (multidrug-resistant), CamWT-C580Y (ART-resistant) [1] | Provide diverse genetic backgrounds for screening against resistant parasites |
| Cell Culture Reagents | RPMI 1640 with Albumax I, hypoxanthine, gentamicin [1] | Support in vitro parasite growth during phenotypic screens |
| Staining Dyes | SYBR Green I, Hoechst 33342, wheat germ agglutinin-Alexa Fluor conjugates [4] [1] | Enable quantification of parasite proliferation and staging |
| Screening Platforms | 384-well and 1536-well plates, automated liquid handlers [4] | Facilitate high-throughput screening of large compound libraries |
| Detection Instruments | High-content imagers (Operetta CLS), flow cytometers, plate readers [1] | Measure assay endpoints and quantify compound effects |
| Compound Libraries | FDA-approved drug libraries, diversity-oriented synthesis libraries, target-focused libraries [1] [56] | Source of chemical starting points for screening campaigns |
| Target Protein Reagents | Recombinant PfATP4, PfDHODH, PfCyt bc1 complex [55] | Enable target-based screening and mechanism validation |
| Proteomic Tools | Affinity resins, mass spectrometry systems, thermal shift assay reagents [55] | Facilitate target deconvolution for phenotypic hits |
Integrated Approaches and Future Directions
The distinction between phenotypic and target-based screening is increasingly blurred with the emergence of integrated approaches that combine strengths of both paradigms. Modern antimalarial discovery often begins with phenotypic screening to identify novel chemotypes, followed by target deconvolution to determine mechanisms of action, and finally structure-based optimization to improve potency and properties [55] [54].
Advanced computational methods are now enhancing phenotypic screening efficiency. The DrugReflector platform uses active reinforcement learning trained on compound-induced transcriptomic signatures to predict compounds that induce desired phenotypic changes, providing an order of magnitude improvement in hit rates compared to random library screening [57]. This approach enables phenotypic screening campaigns to be smaller and more focused while maintaining the biological relevance of phenotypic assessment.
Another emerging trend is the use of highly selective tool compounds for preliminary target deconvolution during phenotypic screening. By screening with compounds of known high selectivity, researchers can immediately associate specific phenotypes with target modulation, accelerating the target identification process [56]. This strategy leverages the growing wealth of bioactivity data in databases like ChEMBL to select optimal tool compounds for phenotypic screening [56].
Figure 3: Integrated drug discovery workflow combining phenotypic and target-based approaches.
Both phenotypic and target-based screening approaches offer distinct advantages and limitations for antimalarial drug discovery. Phenotypic screening excels at identifying novel chemotypes with whole-parasite activity and has historically delivered first-in-class antimalarials, but requires challenging target deconvolution efforts. Target-based screening enables rational optimization and streamlined development but depends on pre-validated targets and may not guarantee whole-parasite activity. The most productive strategy for discovering novel antimalarials with new mechanisms of action involves integrating both approaches, leveraging their complementary strengths to address the urgent need for new therapies against drug-resistant malaria parasites.
The Role of 'Omics and AI in Shaping Next-Generation Screening
The fight against malaria is at a critical juncture. The emergence of Plasmodium falciparum resistance to front-line artemisinin-based combination therapies (ACTs) underscores an urgent need for new antimalarials with novel mechanisms of action [58] [5]. The drug discovery landscape is dominated by two complementary approaches: phenotypic screening, which identifies compounds based on whole-parasite growth inhibition without prior knowledge of molecular targets, and target-based screening, which tests compounds against specific validated molecular targets [4] [12]. Historically, phenotypic screening has delivered the majority of new antimalarial lead compounds discovered over the past decade, largely because it identifies compounds that are already cell-permeable and biologically active in the complex parasite environment [5]. However, target-based approaches offer distinct advantages for downstream optimization, including the ability to develop target-specific biochemical assays and utilize structural information for rational drug design [12].
The integration of 'omics technologies (genomics, transcriptomics, proteomics, metabolomics) with artificial intelligence is now transforming both paradigms, enabling more predictive compound selection, accelerating target identification, and facilitating the discovery of multi-stage inhibitors essential for malaria eradication campaigns [59] [24]. This review compares the evolving capabilities of phenotypic and target-based screening within modern antimalarial discovery, examining how 'omics and AI are bridging historical divisions between these approaches to deliver next-generation therapeutics.
Technological Foundations: Multi-Omics Integration Methodologies
The power of modern screening approaches lies in their ability to integrate multiple layers of biological information. Multi-omics integration strategies provide a comprehensive view of biological systems by connecting upstream genetic determinants with downstream functional outcomes [60] [61].
Table 1: Multi-Omics Integration Approaches in Antimalarial Research
| Integration Approach | Methodology | Application in Antimalarial Research |
|---|---|---|
| Correlation-Based | Gene co-expression analysis with metabolomics | Identifying metabolic pathways co-regulated with gene modules during parasite development [60] |
| Network-Based | Gene-metabolite interaction networks | Mapping regulatory networks between parasite genes and metabolic outputs in response to drug pressure [60] |
| Machine Learning | Multi-modal AI algorithms | Predicting compound activity across parasite life stages by integrating structural and omics data [24] |
| Pathway Integration | Concurrent transcriptomics, proteomics, and metabolomics | Holistic view of drug mechanisms by connecting gene expression changes to functional protein and metabolic outcomes [61] |
The biological insights gained from these integration approaches are transforming how researchers interpret screening results. For instance, concurrent transcriptome, proteome, and metabolome analysis enables researchers to move beyond simple inhibition metrics to understand systems-level responses to compound treatment, connecting gene expression changes to functional protein activities and ultimate metabolic consequences [61]. This is particularly valuable for understanding complex drug mechanisms in the malaria parasite, where polypharmacology (drugs acting on multiple targets) may contribute to efficacy and resistance profiles.
Visualizing Multi-Omics Integration
The following diagram illustrates the conceptual workflow for integrating multi-omics data in antimalarial drug discovery:
Phenotypic Screening: Enhanced by AI and Omics
Phenotypic screening has been responsible for the majority of novel antimalarial compounds discovered in the past decade, with several advancing to clinical development [5]. The traditional strength of phenotypic approaches lies in their ability to identify compounds active against the whole parasite without requiring prior target knowledge, naturally selecting for cell-permeable compounds with relevant biological activity [4].
Recent Advances in Multi-Stage Phenotypic Screening
Contemporary phenotypic screening has evolved beyond simple asexual blood stage inhibition to encompass multiple parasite life cycle stages, addressing the need for compounds that can treat acute disease, prevent relapse, and block transmission:
MMV030084: This trisubstituted imidazole identified through phenotypic screening displays nanomolar potency against asexual blood stages (TCP-1), sexual blood stages (TCP-5), and liver stages (TCP-4) of Plasmodium parasites. Target deconvolution revealed it inhibits P. falciparum cGMP-dependent protein kinase (PfPKG), a key regulator of parasite egress from host cells [5].
WM382: This dual inhibitor of plasmepsin IX and X (PMIX and PMX) prevents merozoite egress and invasion. It demonstrates activity against asexual blood stages, reduces liver stage merozoite viability, and inhibits oocyst development, exhibiting suppressive prophylactic and transmission-blocking potential alongside blood-stage efficacy [5].
Table 2: Recently Discovered Multi-Stage Antimalarial Compounds from Phenotypic Screening
| Compound | Asexual Blood Stage (TCP-1) | Liver Stage (TCP-4) | Sexual Stage (TCP-5) | Identified Molecular Target |
|---|---|---|---|---|
| MMV030084 | IC50 ~100nM | Active (P. berghei) | Active | PfPKG [5] |
| WM382 | IC50 <100nM | Active | Active (oocyst) | Plasmepsin IX/X [5] |
| SGI-1027 | Low-nanomolar activity | Not reported | Active (early gametocytes) | DNA methyltransferase [5] |
AI-Enhanced Phenotypic Screening Platforms
Artificial intelligence has dramatically accelerated phenotypic screening by enabling virtual compound prioritization. Platforms like MalariaFlow represent the cutting edge of this approach, offering comprehensive deep learning capabilities for predicting antimalarial activity across multiple Plasmodium life stages and strains [24]. This platform systematically compares multiple AI algorithms (including Random Forest, XGBoost, Graph Neural Networks, and Transformer-based models) trained on over 410,000 experimentally validated compounds with activity annotations across ten phenotypic characteristics and three major life cycle stages [24].
The DeepMalaria platform demonstrates how deep learning can enhance hit identification, using graph convolutional neural networks (GCNNs) to predict anti-Plasmodium inhibitory properties directly from compound structures (SMILES) [62]. When applied to a macrocyclic compound library, DeepMalaria successfully identified all compounds with nanomolar activity and 87.5% of compounds with >50% inhibition, significantly improving screening efficiency [62].
Experimental Protocol: In Vitro Phenotypic Screening
Standardized experimental protocols are essential for generating high-quality phenotypic screening data:
Parasite Culture: Maintain P. falciparum parasites (typically drug-sensitive 3D7 and multidrug-resistant Dd2 strains) in human erythrocytes at 2% hematocrit in complete RPMI-1640 medium with 0.5% Albumax under mixed gas conditions (90% N2, 5% O2, 5% CO2) [62].
Compound Treatment: Dispense compounds in serial dilutions across 384-well plates. Include controls (untreated parasites, 100% inhibition controls).
Viability Assessment: After 72-hour incubation, measure parasite viability using the SYBR Green I fluorescence assay. This DNA-binding dye produces fluorescence proportional to parasite DNA content [62].
Data Analysis: Calculate percentage inhibition relative to controls, determine IC50 values using nonlinear regression, and apply machine learning models for compound prioritization.
Target-Based Screening: Leveraging Omics for Validation
While phenotypic screening identifies bioactive compounds, target-based approaches focus on specific molecular targets, offering advantages for rational drug design and optimization. The shift toward target-based antimalarial discovery has been enabled by the identification and validation of molecular targets through multi-omics approaches [12].
Omics-Driven Target Identification and Prioritization
Genomic and genetic approaches have empowered novel methods for studying gene function in Plasmodium falciparum. Several techniques are now routinely used for target identification and validation:
Resistance Generation with Whole-Genome Sequencing: In vitro selection of drug-resistant parasites followed by whole-genome sequencing identifies mutations that may indicate the molecular target or resistance mechanisms [12].
Thermal Proteome Profiling: This mass-spectrometry-facilitated approach monitors drug-induced thermal stability shifts in proteins to identify putative targets without needing to generate resistant parasites [12].
Metabolomic Profiling: Analysis of metabolic changes in drug-treated parasites provides fingerprints of compounds acting through known mechanisms and can suggest novel modes of action [12].
The Malaria Drug Accelerator (MalDA) consortium has established criteria for prioritizing antimalarial drug targets, emphasizing targets with strong genetic validation, essentiality across multiple life cycle stages, and low potential for resistance development [12].
Experimental Protocol: Target-Based Screening
Target-based screening employs different experimental workflows focused on molecular interactions:
Target Selection and Validation: Select molecular targets based on genetic evidence (e.g., gene knockout studies), essentiality data, and druggability assessment. Priority targets include Plasmodium cGMP-dependent protein kinase (PfPKG), plasmepsins IX and X, and various amino acyl tRNA synthetases [12] [5].
Biochemical Assay Development: Develop purified enzyme assays with appropriate detection methods (fluorescence, luminescence, etc.) suitable for high-throughput screening.
Compound Screening: Screen compound libraries against the validated target, typically testing compounds at 10-20μM in concentration-response formats.
Counter-Screening: Test hits against human orthologs (if present) to assess selectivity and against mammalian cell lines (e.g., HepG2) to assess cytotoxicity [62].
Cellular Validation: Confirm cellular activity of biochemical hits using whole-cell phenotypic assays against Plasmodium blood stages.
Comparative Analysis: Phenotypic vs. Target-Based Screening
The integration of omics and AI is blurring the historical divisions between phenotypic and target-based screening approaches. The following comparison highlights their complementary strengths:
Table 3: Direct Comparison of Phenotypic and Target-Based Screening Approaches
| Parameter | Phenotypic Screening | Target-Based Screening |
|---|---|---|
| Hit Identification Rate | ~0.5-1% from diverse compound libraries [4] | Varies significantly by target class and library design |
| Target Deconvolution Requirement | Required (can be challenging) [12] | Not required (target known from outset) |
| Chemical Starting Points | Identifies novel chemotypes with whole-cell activity [5] | May identify hits with insufficient permeability for cellular activity |
| Multi-Stage Activity Assessment | Compatible with specialized assays for liver, blood, and sexual stages [24] | Requires separate target validation for each life stage |
| Optimization Complexity | Can be challenging without target knowledge [12] | More straightforward with structural information and biochemical assays |
| Resistance Prediction | Limited without target knowledge | Enabled by target understanding and mutational analysis |
Integrated Screening Workflows: The Future of Antimalarial Discovery
The most advanced antimalarial discovery programs now combine phenotypic and target-based approaches in integrated workflows that leverage the strengths of both paradigms. The following diagram illustrates this synergistic relationship:
This integrated approach creates a virtuous cycle where phenotypic screening identifies novel chemotypes with whole-cell activity, multi-omics approaches elucidate their mechanisms of action, and target-based screening enables optimization of selective inhibitors against validated targets. AI serves as the connective tissue throughout this process, predicting compound properties, identifying structure-activity relationships, and prioritizing compounds for synthesis and testing [59] [24].
Essential Research Toolkit
Implementation of modern screening approaches requires specialized reagents and computational tools:
Table 4: Essential Research Reagents and Platforms for Next-Generation Screening
| Tool/Category | Specific Examples | Research Application |
|---|---|---|
| AI Screening Platforms | MalariaFlow [24], DeepMalaria [62] | Virtual compound screening and activity prediction across life stages |
| Target Identification Methods | Thermal Proteome Profiling [12], Resistance generation + WGS [12] | Identifying molecular targets of phenotypic hits |
| Multi-Omics Integration | Gene-metabolite networks [60], Co-expression analysis [60] | Understanding system-wide drug effects |
| Phenotypic Assay Systems | SYBR Green I fluorescence assay [62], Luciferase reporter assays [4] | Measuring parasite viability and compound inhibition |
| Chemical Libraries | Macrocyclic compounds [62], Natural product-inspired libraries [62] | Source of novel chemotypes with antimalarial activity |
The integration of 'omics technologies and artificial intelligence is transforming both phenotypic and target-based screening paradigms in antimalarial research. Rather than competing approaches, they have become complementary components of integrated discovery workflows. Phenotypic screening continues to identify novel chemotypes with desirable whole-cell activity properties, while target-based approaches facilitate rational optimization through structural biology and mechanistic understanding. Multi-omics integration provides the connective framework that unites these approaches, offering systems-level insights into drug mechanisms and parasite biology. As AI algorithms become increasingly sophisticated and multi-omics datasets expand, the distinction between phenotypic and target-based screening will continue to blur, ultimately accelerating the delivery of novel antimalarials capable of addressing the urgent threat of drug-resistant malaria.
The relentless burden of malaria, compounded by the emergence and spread of partial resistance to artemisinin and partner drugs, underscores the urgent need for a diversified portfolio of new antimalarials with novel modes of action (MoA) [63] [13]. Over the past two decades, antimalarial drug discovery has been propelled by two primary screening paradigms: phenotypic screening, which evaluates compound activity against the whole parasite in its cellular context, and target-based screening, which investigates the effect of compounds on specific, purified molecular targets [7] [4]. A recent review of the clinical candidates delivered between 2005 and 2025 concluded that the phenotypic-based approach has outperformed the target-based approach in antimalarial drug discovery [7]. This comparative success is largely because phenotypic screens do not require prior knowledge of a drug target, inherently select for cell-permeable compounds, and can identify agents with cooperative effects or those acting on multiple targets [4].
However, the future of antimalarial research does not lie in the exclusive use of one paradigm over the other, but in their strategic integration. The next generation of drug discovery workflows is evolving into a unified, AI-powered pipeline that synergistically combines the breadth of phenotypic screening with the precision of target-based methods. This guide objectively compares the current state of these approaches and details the experimental protocols and technological advancements—particularly in artificial intelligence (AI) and machine learning (ML)—that are forging a path toward a fully integrated, more efficient future for antimalarial drug development.
Comparative Analysis of Screening Paradigms
Performance and Clinical Output
The table below summarizes the key characteristics and outcomes of the phenotypic and target-based approaches as applied to antimalarial discovery.
Table 1: Comparison of Phenotypic vs. Target-Based Screening in Antimalarial Discovery
| Aspect | Phenotypic Screening | Target-Based Screening |
|---|---|---|
| Fundamental Principle | Measures compound-induced changes in whole parasites or specific lifecycle stages [4]. | Measures compound effects on a specific, purified protein or molecular target [4] [1]. |
| Key Advantage | No need for prior target knowledge; identifies cell-permeable compounds; can reveal multi-target or novel MoAs [4]. | High specificity; allows for rational, structure-based drug design [13]. |
| Primary Limitation | MoA and specific target are initially unknown, requiring subsequent deconvolution efforts [4]. | Hits may not show activity in whole-cell assays due to permeability or metabolic issues [4] [24]. |
| Notable Clinical Output | Has delivered multiple clinical candidates in the last 20 years, including KAE609, KAF156, and M5717 [7] [24]. | Clinical output has been more limited compared to the phenotypic approach in the antimalarial field [7]. |
Methodologies and Experimental Protocols
Protocol for Image-Based Phenotypic High-Throughput Screening (HTS)
This protocol, adapted from a 2025 HTS and meta-analysis study, details a modern, image-based phenotypic screen against asexual blood stage (ABS) Plasmodium falciparum [1].
Step 1: Compound Library and Plate Preparation An in-house library of 9,547 small molecules was used. Stock compounds were prepared in 100% DMSO and then diluted in phosphate-buffered saline (PBS). Using automated liquid handlers (e.g., Hummingwell), compounds were transferred into 384-well glass plates for screening.
Step 2: Parasite Culture and Synchronization P. falciparum parasites (e.g., strain 3D7) are maintained in vitro in O+ human red blood cells (RBCs) using complete RPMI 1640 culture medium. To ensure synchronicity, parasites are treated with 5% sorbitol, which lyses later-stage parasites, resulting in a highly synchronized population of ring stages.
Step 3: Drug Sensitivity Assay Synchronized schizont-stage parasites at 1% parasitemia and 2% hematocrit are dispensed into the compound-treated 384-well plates. The plates are incubated for 72 hours in a specialized malaria culture chamber at 37°C with a mixed gas environment (1% O₂, 5% CO₂ in N₂).
Step 4: Staining and Image Acquisition After incubation, the assay plate is diluted to 0.02% hematocrit and transferred to a specialized 384-well microplate. The cells are stained and fixed with a solution containing:
- Wheat germ agglutinin–Alexa Fluor 488: Binds to glycoproteins on the RBC membrane, outlining all cells.
- Hoechst 33342: A DNA-intercalating dye that stains parasite nuclei.
- Paraformaldehyde: Fixes the cells, preserving morphological details. After a 20-minute incubation, nine image fields per well are acquired using a high-content imaging system (e.g., Operetta CLS) with a 40x water immersion lens.
Step 5: Image Analysis and Hit Identification The acquired images are analyzed using specialized software (e.g., Columbus). The algorithm identifies and segments individual RBCs based on the Alexa Fluor 488 signal and then detects the presence of parasitic DNA within those cells via the Hoechst signal. This allows for the precise quantification of parasitemia in each well. Compounds that significantly reduce parasitemia compared to control wells are identified as "hits." In the referenced study, the top 3% of actives (256 compounds) were selected for further dose-response analysis to determine their half-maximal inhibitory concentration (IC₅₀) [1].
Protocol for a Target-Based Screen
While a specific protocol for a malaria target-based screen was not detailed in the provided results, the general workflow is well-established.
Step 1: Target Selection and Protein Production A putative essential protein from Plasmodium (e.g., an enzyme involved in metabolic pathways) is identified and validated as a drug target. The protein is then cloned, expressed, and purified to homogeneity.
Step 2: Assay Development A biochemical assay is developed to measure the activity of the purified protein. This could be a fluorescence-based, absorbance-based, or radiometric assay that produces a signal proportional to the enzyme's activity.
Step 3: High-Throughput Screening The purified target protein is incubated with compounds from a library in microtiter plates. The biochemical reaction is initiated by adding the substrate, and the signal is measured after a fixed time. Compounds that significantly reduce the signal output are identified as hits for the specific molecular target.
Step 4: Counter-Screening and Validation Hits from the biochemical assay must be tested in whole-cell phenotypic assays to confirm they can penetrate the parasite and exert the desired antimalarial effect. This critical step often represents a major bottleneck, as many potent enzyme inhibitors fail at this stage [4] [24].
The Integration of AI and Novel Technologies
The distinction between phenotypic and target-based screening is becoming increasingly blurred by the integration of AI and machine learning, which creates a feedback loop that enhances both approaches.
Table 2: Emerging AI Platforms and Their Applications in Antimalarial Drug Discovery
| Technology / Platform | Type | Primary Application | Key Benefit |
|---|---|---|---|
| MalariaFlow [24] | Deep Learning Platform | Predicts antimalarial activity across 10 Plasmodium phenotypes and 3 lifecycle stages. | Identifies multi-stage inhibitors; outperformed classical ML and other DL models (AUROC of 0.900). |
| MMV/LPIXEL/Univ. Dundee AI Partnership [63] | AI-Powered Image Analysis | Uses "cell painting" and ML pattern recognition to understand a compound's MoA from images. | Dramatically accelerates MoA deconvolution, saving months of experimental work. |
| CDD Vault [9] | Integrated AI Platform | Provides collaborative data management with integrated AI tools for predictive modeling and virtual screening. | Democratizes access to advanced AI for researchers in resource-limited settings. |
AI models, particularly co-representation models like FP-GNN that fuse molecular graph structures with chemical domain knowledge, have demonstrated superior performance in predicting antimalarial activity [24]. These models can be trained on massive, manually curated datasets containing hundreds of thousands of bioactivity data points across multiple parasite lifecycle stages and resistant strains. This allows for the in silico prediction of compound activity and prioritization before laboratory testing, making the screening process faster and more cost-effective [24].
Furthermore, AI is revolutionizing the interpretation of phenotypic screens. Traditionally, a major bottleneck of phenotypic screening has been the lengthy process of MoA deconvolution. A new partnership between MMV, LPIXEL, and the University of Dundee aims to solve this by using AI-powered image analysis. This technology uses machine learning pattern recognition on images of stained parasite cells (a process known as cell painting) to provide immediate insights into a compound's biological impact and its likely MoA, a task that previously took months [63].
Visualizing the Evolving Workflow
The following diagrams illustrate the transition from the traditional, siloed screening approaches to a modern, AI-integrated workflow.
Traditional Siloed Screening Approaches
Traditional Screening Workflow
An Integrated, AI-Powered Discovery Pipeline
AI-Integrated Screening Workflow
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful implementation of an integrated drug discovery pipeline relies on a suite of essential reagents and tools. The table below lists key solutions used in the featured experiments.
Table 3: Key Research Reagent Solutions for Antimalarial Screening
| Reagent / Solution | Function in the Workflow | Example Use Case |
|---|---|---|
| Synchronized P. falciparum Cultures | Provides a homogeneous population of parasites at a specific lifecycle stage for consistent and reproducible screening. | Essential for all phenotypic screens (ABS, gametocyte) to accurately assess stage-specific compound activity [1] [23]. |
| Transgenic Parasite Lines (e.g., NF54/iGP1_RE9Hulg8) | Engineered to express viability reporters (e.g., luciferase); enables conditional production of specific stages and highly sensitive quantification of parasite burden. | Used in advanced transmission-blocking platforms for in vitro and in vivo gametocyte screening [23]. |
| Fluorescent Probes (Hoechst 33342, WGA-Alexa Fluor 488) | Enable high-content imaging by specifically staining parasite DNA (Hoechst) and erythrocyte membranes (WGA). | Critical for image-based HTS to automatically quantify parasitemia and assess parasite morphology [1]. |
| In-house or Collaborative Compound Libraries | Curated collections of small molecules, including FDA-approved drugs and diverse chemical scaffolds, which serve as the starting point for discovery. | Screened to identify initial "hit" compounds with antimalarial activity [1]. |
| AI/ML Prediction Platforms (e.g., MalariaFlow) | Web servers that use trained deep learning models to predict antimalarial activity and enable virtual screening. | Used for in silico prioritization of compounds from vast virtual libraries before synthesis or purchasing, accelerating hit discovery [24]. |
The historical comparison clearly demonstrates the empirical success of phenotypic screening in delivering new antimalarial clinical candidates over the last two decades. However, the future of malaria drug discovery is not a choice between phenotypic and target-based approaches but hinges on their strategic fusion. The emerging paradigm is a fully integrated, AI-driven workflow that leverages the target-agnostic, physiologically relevant power of phenotypic screening and augments it with AI-powered MoA prediction, target deconvolution, and in silico optimization. This synergistic integration, exemplified by platforms like MalariaFlow and new AI-powered image analysis partnerships, is poised to dramatically accelerate the discovery of the next generation of multi-stage antimalarial drugs, bringing us closer to the ultimate goal of malaria eradication.
Conclusion
The strategic competition between phenotypic and target-based screening has profoundly enriched the antimalarial drug discovery landscape. Evidence confirms phenotypic screening's superior track record in delivering first-in-class clinical candidates by exploring novel biological space without target bias. However, the future lies not in choosing one approach over the other, but in their intelligent integration. Target-based methods are being revitalized by chemically validated targets from phenotypic hits, while phenotypic assays are becoming more sophisticated with complex disease models. For researchers, the key is to leverage the strengths of each: using phenotypic screening to identify novel mechanisms and target-based approaches to rationally optimize selective compounds. The continued adoption of functional genomics, advanced bioinformatics, and machine learning will further blur the lines between these paradigms, creating a more holistic and efficient pipeline. Ultimately, defeating malaria will require this synergistic strategy, combining the serendipitous discovery power of phenotypics with the precision engineering of target-based design to develop the multi-stage, resistance-proof antimalarials of tomorrow.
References
- 1. High-throughput screening and meta-analysis for lead ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590855/]
- 2. The evolving threat of drug-resistant malaria in a ... [https://www.sciencedirect.com/science/article/abs/pii/S0924857925001980]
- 3. Understanding the global rise of artemisinin resistance [https://elifesciences.org/articles/105544]
- 4. Phenotypic screens in antimalarial drug discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5007148/]
- 5. Promising antimalarial hits from phenotypic screens [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2023.1308193/full]
- 6. A decade of malaria phenotypic screenings: Key lessons ... [https://www.sciencedirect.com/science/chapter/bookseries/abs/pii/S0065774319300120]
- 7. versus target-based approaches in antimalarial drug ... [https://pubmed.ncbi.nlm.nih.gov/41178292/]
- 8. Target Identification vs. Phenotypic Screening in Drug ... [https://pharmafeatures.com/strategic-dichotomy-target-identification-vs-phenotypic-screening-in-drug-discovery/]
- 9. Advancements in Antimalarial Drug Discovery and ... [https://www.collaborativedrug.com/cdd-blog/advancements-in-antimalarial-drug-discovery-and-development]
- 10. PBPK-Led Assessment of Antimalarial Drug ... [https://pubmed.ncbi.nlm.nih.gov/39930940/]
- 11. Physiologically based pharmacokinetic modelling to predict ... [https://tropmedhealth.biomedcentral.com/articles/10.1186/s41182-025-00790-w]
- 12. Prioritization of Molecular Targets for Antimalarial Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8608365/]
- 13. Antimalarial drug resistance and drug discovery: learning ... [https://www.sciencedirect.com/science/article/pii/S2211320725000259]
- 14. High-throughput screening and meta-analysis for lead ... [https://malariajournal.biomedcentral.com/articles/10.1186/s12936-025-05587-0]
- 15. Target-Based Virtual Screening of Natural Compounds ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2022.850176/full]
- 16. Target Product Profiles & Target Candidate Profiles [https://www.mmv.org/research-development/information-scientists/target-product-profiles-target-candidate-profiles]
- 17. DPDx - Malaria [https://www.cdc.gov/dpdx/malaria/index.html]
- 18. Plasmodium—a brief introduction to the parasites causing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7792015/]
- 19. Phenotypic Versus Target-Based Screening for Drug ... [https://www.technologynetworks.com/drug-discovery/articles/phenotypic-versus-target-based-screening-for-drug-discovery-300037]
- 20. Opportunities and challenges in phenotypic drug discovery [https://www.nature.com/articles/nrd.2017.111]
- 21. Phenotypic vs. target-based drug discovery for first-in-class ... [https://pubmed.ncbi.nlm.nih.gov/23511784/]
- 22. New Insights into Malaria Could Reshape Treatment [https://www.cuimc.columbia.edu/news/new-insights-malaria-could-reshape-treatment]
- 23. An all-in-one pipeline for the in vitro discovery and in vivo ... [https://www.nature.com/articles/s41467-025-62014-3]
- 24. MalariaFlow: A comprehensive deep learning platform for ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523424006573]
- 25. CRISPRi-based phenotypic screening in bacteria [https://link.springer.com/article/10.1007/s12257-025-00241-7]
- 26. Assessment In Vitro of the Antimalarial and Transmission ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8923224/]
- 27. Chemoprotective antimalarials identified through ... [https://www.nature.com/articles/s41598-021-81486-z]
- 28. from chemical biology to first-in-class drugs [https://link.springer.com/article/10.1007/s12272-015-0645-0]
- 29. Cipargamin - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/cipargamin]
- 30. Comparative chemical genomics reveal that the ... [https://www.nature.com/articles/srep27806]
- 31. Novartis - KAF156 Molecule [https://www.amrindustryalliance.org/case-study/kaf156-molecule/]
- 32. Ganaplacide - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/medicine-and-dentistry/ganaplacide]
- 33. Novartis and Medicines for Malaria Venture launch patient ... [https://www.novartis.com/news/media-releases/novartis-and-medicines-malaria-venture-launch-patient-trial-africa-kaf156-novel-compound-against-multidrug-resistant-malaria]
- 34. Inhibition of Resistance-Refractory P. falciparum Kinase PKG ... [https://pubmed.ncbi.nlm.nih.gov/32359426/]
- 35. New targets for antimalarial drug discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9934905/]
- 36. Genome-wide functional screening of drug-resistance ... [https://www.nature.com/articles/s41467-022-33804-w]
- 37. Structural comparison of human and Plasmodium proteasome ... [https://malariajournal.biomedcentral.com/articles/10.1186/s12936-025-05259-z]
- 38. Integrated structural biology of the native malarial ... [https://www.nature.com/articles/s41594-025-01632-3]
- 39. A structural comparative approach to identifying novel ... [https://www.sciencedirect.com/science/article/abs/pii/S1476927113000364]
- 40. Review Phenotypic Screens in Antimalarial Drug Discovery [https://www.sciencedirect.com/science/article/abs/pii/S1471492216300319]
- 41. Computational identification of aspartic protease inhibitors ... [https://www.nature.com/articles/s41598-025-98516-9]
- 42. The Deconvolution Revolution: Strategies for Target ... [https://momentum.bio/content/the-deconvolution-revolution-strategies-for-target-identification/]
- 43. Target deconvolution techniques in modern phenotypic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3594516/]
- 44. Phenotypic profiling, Ardigen phenAID [https://ardigen.com/phenotypic-profiling-ardigen-phenaid/]
- 45. Advances in omics-based methods to identify novel targets for malaria and other parasitic protozoan infections - PubMed [https://pubmed.ncbi.nlm.nih.gov/31640748/]
- 46. Decoding phenotypic screening: A comparative analysis of ... [https://www.sciencedirect.com/science/article/pii/S2001037024000461]
- 47. Changes in susceptibility of Plasmodium falciparum to ... [https://www.nature.com/articles/s41467-025-62810-x]
- 48. Hit Validation [https://spirochem.com/discovery/medicinal-chemistry/hit-validation]
- 49. Top 5 Drug Discovery Trends 2025 Driving Breakthroughs [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 50. High-throughput screening and meta-analysis for lead ... [https://pubmed.ncbi.nlm.nih.gov/41199304/]
- 51. Antimalarial drug discovery: progress and approaches - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10543600/]
- 52. Novartis Phase III trial for next-generation malaria ... [https://www.novartis.com/news/media-releases/novartis-phase-iii-trial-next-generation-malaria-treatment-klu156-ganlum-meets-primary-endpoint-potential-combat-antimalarial-resistance]
- 53. Quantifying the pharmacology of antimalarial drug ... [https://www.nature.com/articles/srep32762]
- 54. Phenotypic and targeted drug discovery in immune therapeutics [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d5ra03914b]
- 55. Current and emerging target identification methods for ... [https://www.sciencedirect.com/science/article/pii/S2211320722000288]
- 56. A data-driven journey using results from target-based drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12062751/]
- 57. Improving phenotypic screening | Nature Chemical Biology [https://www.nature.com/articles/s41589-025-02093-x]
- 58. Genomic and Genetic Approaches to Studying Antimalarial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8162148/]
- 59. Insights from NextGen Omics - Ardigen | Top AI-Powered CRO... 2025 [https://ardigen.com/insights-from-nextgen-omics-2025/]
- 60. Strategies for Comprehensive Multi-Omics Integrative Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11592251/]
- 61. Transcriptomics + Proteomics + Metabolomics [https://www.metwarebio.com/transcriptomics-proteomics-metabolomics/]
- 62. DeepMalaria: Artificial Intelligence Driven Discovery of Potent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6974622/]
- 63. MMV announces new partnership using AI-powered image ... [https://www.mmv.org/newsroom/news-resources-search/mmv-announces-new-partnership-using-ai-powered-image-analysis]