Advancing Quality Control in Protozoan Microscopy: From Foundational Methods to AI-Powered Solutions
This article provides a comprehensive overview of quality control in the microscopic identification of protozoan parasites, a critical process for research and drug development.
Advancing Quality Control in Protozoan Microscopy: From Foundational Methods to AI-Powered Solutions
Abstract
This article provides a comprehensive overview of quality control in the microscopic identification of protozoan parasites, a critical process for research and drug development. It explores the foundational challenges of traditional microscopy, including operator dependency and low throughput. The content details the integration of advanced methodologies, such as automated digital microscopy and convolutional neural networks (CNNs), which enhance detection accuracy and workflow efficiency. It further addresses common troubleshooting scenarios and optimization strategies for sample processing and DNA extraction. Finally, the article presents a rigorous validation framework for these new technologies, comparing their performance against gold-standard methods and discussing their implications for improving diagnostic yield and accelerating biomedical discovery.
The Evolving Landscape of Protozoan Diagnostics: Challenges and Imperatives for Quality Control
The Global Burden of Protozoan Infections and the Critical Role of Accurate Diagnosis
Protozoan pathogens represent a significant and ongoing threat to global public health, contributing substantially to diarrheal morbidity and mortality worldwide, particularly in resource-limited settings [1]. A recent systematic review and meta-analysis covering studies from 1999 to 2024 revealed a global protozoan prevalence of 7.5% (95% CI: 5.6%-10.0%) in diarrheal cases, with the highest burden observed in the Americas and Africa [1]. Among these pathogens, Giardia and Cryptosporidium species were identified as the most common causative agents [1].
The economic impact of parasitic infections extends deeply into healthcare systems and developing economies. India alone spends approximately 0.34% of its total consumption expenditure on infectious diseases including parasitic infections, with malaria control costing the country US$1,940 million in 2014 [2]. Beyond human health, protozoan infections seriously affect livestock and agriculture, with plant-parasitic nematodes causing global crop yield losses estimated at $125 to $350 billion annually [3].
Table 1: Global Burden of Major Protozoan Infections
| Pathogen/Disease | Estimated Cases/Impact | Key Endemic Regions | Primary Population Affected |
|---|---|---|---|
| Malaria (Plasmodium spp.) | 249 million cases, >600,000 deaths annually [3] | Tropical regions globally | Children under 5 years (80% of deaths) |
| Diarrheal Protozoa | 7.5% prevalence in diarrheal cases [1] | Americas, Africa | All age groups, higher burden in resource-limited settings |
| Visceral Leishmaniasis | Up to 400,000 new cases annually [3] | Brazil, India, East Africa, Southern Europe | Children and young adults |
| Toxoplasmosis (T. gondii) | Up to 1/3 of global population infected [3] | Worldwide | Immunocompromised individuals, pregnant women |
The Diagnostic Journey: From Microscopy to Artificial Intelligence
The evolution of parasitic diagnosis represents a journey from basic morphological identification to sophisticated technologies that enhance detection accuracy and efficiency [2].
Historical Foundations: The Microscopic Era
The 17th century marked a pivotal moment in parasitology with Antonie van Leeuwenhoek's invention of the microscope, which first enabled researchers to visualize the intricate forms of parasites [2]. For centuries, microscopy remained the cornerstone of parasitic diagnosis, with various staining techniques developed to enhance morphological differentiation. Before this technological advancement, parasitic infections were often poorly understood and misdiagnosed, with symptoms frequently attributed to supernatural forces or bodily imbalances [2].
Modern Diagnostic Challenges and Solutions
Despite technological advancements, traditional microscopy maintains both relevance and limitations in contemporary diagnostic practice. The technique remains widely accessible but faces challenges in sensitivity, specificity, and requirement for expert interpretation [2].
Table 2: Evolution of Diagnostic Modalities in Parasitology
| Diagnostic Era | Technologies/Methods | Key Advantages | Principal Limitations |
|---|---|---|---|
| Microscopic Era (17th century - present) | Light microscopy, Staining techniques (e.g., Gram, acid-fast) | Accessibility, Low cost, Direct pathogen visualization | Low sensitivity, Requires expertise, Subjective interpretation |
| Serological Era (20th century - present) | ELISA, Immunoblot, Rapid diagnostic tests | Higher throughput, Detects immune response | Cross-reactivity, Cannot distinguish past/current infection [2] |
| Molecular Era (21st century - present) | PCR, Multiplex assays, Next-generation sequencing | High sensitivity/specificity, Strain differentiation, Quantification | Cost, Infrastructure requirements, Technical expertise [2] |
| AI-Integrated Era (Emerging) | Convolutional neural networks, Deep learning, Automated image analysis | Enhanced accuracy, High-throughput screening, Reduced subjectivity | Requires diverse training datasets, Computational resources [2] |
Technical Support Center: Troubleshooting Guides and FAQs
Frequently Asked Questions: Diagnostic Challenges
Q1: What are the most common causes of false-negative results in microscopic identification of intestinal protozoa?
A1: False negatives most frequently result from:
- Inadequate specimen collection: Insufficient sample volume or improper timing
- Irregular parasite shedding: Non-uniform excretion of parasites in stool
- Suboptimal staining: Improper staining technique or expired reagents
- Operator fatigue: Visual fatigue during manual microscopy screening
- Low parasite load: Concentration below the detection threshold of microscopy
Q2: How can our laboratory improve consistency in morphological identification of protozoan cysts and trophozoites?
A2: Implement these quality control measures:
- Establish a reference image library with confirmed specimens
- Implement double-blind reading for difficult cases
- Participate in proficiency testing programs regularly
- Standardize staining protocols across all technicians
- Incorporate regular calibration of microscope optics
Q3: What validation steps should we follow when implementing a new molecular diagnostic assay for protozoan detection?
A3: A comprehensive validation should include:
- Analytical sensitivity (limit of detection) using serial dilutions of reference standards
- Analytical specificity testing against common commensals and phylogenetically related species
- Reproducibility assessment across multiple operators and days
- Clinical validation against a gold standard method on well-characterized clinical samples
- Establishment of internal quality control parameters for routine use
Troubleshooting Guide: Microscopy Image Analysis
Table 3: Common Image Analysis Challenges and Solutions in Protozoan Diagnostics
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| Poor image quality/blurring | Improper focus, Thick specimens, Condenser misalignment | Use standardized mounting techniques, Calibrate microscope regularly, Employ thin smears |
| Weak or uneven staining | Expired reagents, Improper staining time, Inadequate fixation | Freshly prepare staining solutions, Standardize timing, Check pH of buffers |
| Difficulty distinguishing similar structures | Inadequate resolution, Poor contrast, Similar morphologies | Use oil immersion objectives, Employ differential interference contrast, Utilize specific fluorescent labels |
| Debris misinterpreted as parasites | Sample contamination, Excessive background | Improve sample preparation, Use concentration methods, Implement deep learning segmentation [4] |
| Inconsistent measurements across users | Subjective thresholds, Variable segmentation parameters | Standardize analysis protocols, Use automated detection algorithms, Establish clear criteria [4] |
Experimental Protocols for Quality Control
Protocol: Validation of Staining Procedures for Intestinal Protozoa
Principle: This protocol establishes a standardized method for validating staining procedures used in the morphological identification of intestinal protozoa in clinical specimens.
Materials:
- Microscope with 100x oil immersion objective
- Staining reagents (e.g., Trichrome, iodine, modified acid-fast)
- Quality control slides with known positive specimens
- Reference images of target organisms
Procedure:
- Prepare smears from well-characterized positive control specimens
- Apply staining procedure according to established protocol
- Examine stained smears microscopically using standardized illumination
- Compare staining characteristics with reference standards
- Document color, morphological preservation, and clarity of diagnostic features
- Repeat with negative controls to assess specificity
- Establish acceptance criteria for staining quality
Troubleshooting:
- Over-staining: Reduce staining time or concentration
- Under-staining: Increase staining time or check reagent freshness
- Precipitate formation: Filter stains before use
- Poor morphological preservation: Optimize fixation method
Protocol: Implementation of AI-Assisted Microscopy Screening
Principle: This protocol outlines the steps for implementing and validating deep learning algorithms for automated detection of protozoan parasites in stained specimens.
Materials:
- Digitized whole slide images or high-resolution micrographs
- Annotation software for training data generation
- Computational resources for model training
- Diverse dataset representing various parasite stages and species
Procedure:
- Data Curation: Collect and annotate a minimum of 500-1000 images per target parasite
- Data Partitioning: Divide dataset into training (70%), validation (15%), and test (15%) sets
- Model Selection: Choose appropriate neural network architecture (e.g., CNN, U-Net)
- Training: Implement iterative training with data augmentation
- Validation: Assess performance on independent test set
- Integration: Deploy model for assisted screening with human oversight
Quality Control Measures:
- Regular performance monitoring with challenge slides
- Continuous expansion of training dataset with difficult cases
- Maintenance of human expert review for ambiguous cases
- Documentation of algorithm performance metrics over time
Research Reagent Solutions for Protozoan Diagnostics
Table 4: Essential Research Reagents for Protozoan Identification and Characterization
| Reagent/Material | Function/Application | Key Considerations |
|---|---|---|
| Trichrome Stain | Differentiates internal structures of intestinal protozoa | Batch-to-batch variability requires QC; staining time affects clarity |
| Modified Acid-Fast Stain | Identifies Cryptosporidium, Cyclospora, Cystoisospora | Decolorization must be optimized; requires specific safety handling |
| Specific Fluorescent Antibodies | Direct detection of antigens in specimens | Lot validation required; potential cross-reactivity must be assessed |
| PCR Master Mixes | Molecular amplification of parasite DNA | Inhibitor-resistant formulations preferred; validation for multiplexing |
| DNA Extraction Kits | Nucleic acid purification from diverse specimens | Efficiency varies by parasite and specimen type; internal controls critical |
| Reference Genomic DNA | Positive controls for molecular assays | Quantification and purity verification essential; prevent contamination |
| Culture Media | Propagation of specific protozoa for QC strains | Species-specific formulations; strict contamination prevention |
| Protein Quality Control Reagents | Study of parasite stress response mechanisms [5] [6] | Target HSPs, chaperones; potential therapeutic applications |
Workflow Visualization: Diagnostic Pathways
Diagnostic Evolution Pathway
Image Analysis Troubleshooting Pathway
The field of protozoan diagnostics continues to evolve rapidly, with emerging technologies offering promising avenues for addressing current limitations. Artificial intelligence and deep learning approaches, particularly convolutional neural networks, are revolutionizing parasitic diagnostics by enhancing detection accuracy and efficiency [2]. These technologies demonstrate particular promise for overcoming the subjectivity and operator dependency that have long challenged microscopic identification.
Innovative imaging technologies, including three-dimensional label-free optical diffraction holotomography (3D-ODH), are enabling more precise identification of parasite-host interactions through non-invasive, high-resolution imaging [7]. Meanwhile, research continues to advance our understanding of fundamental parasite biology, including protein quality control machinery that may offer new therapeutic targets [5] [6]. As these technologies mature, their integration into standardized diagnostic workflows will be essential for reducing the global burden of protozoan infections through earlier detection, more targeted treatment, and improved surveillance.
Traditional microscopy, while a foundational tool in protozoan research, presents significant challenges for quality control in modern scientific and drug development settings. Three core limitations hinder its reliability and efficiency: operator dependency, subjectivity in interpretation, and inherently low throughput. These issues are particularly problematic in high-stakes environments like pharmaceutical development, where consistency and objectivity are paramount. This guide details these challenges and provides troubleshooting advice, advanced protocols, and resources to help researchers mitigate these constraints.
The table below summarizes the primary limitations and their impact on protozoan research quality control.
| Limitation | Impact on Quality Control | Common Symptoms & Errors |
|---|---|---|
| Operator Dependency [2] [8] | High variability in sample preparation, focusing, and diagnosis compromises reproducibility and data integrity. | - Blurry or out-of-focus images [8]- Inconsistent identification of protozoan species (e.g., misidentifying C. parvum vs. C. hominis) [9]- Uneven illumination across the field of view [8] |
| Subjectivity [2] | Non-standardized visual analysis introduces bias, affecting the accuracy of pathogen identification and morphological characterization. | - Disagreement in parasite counts or staging between technicians [2]- Inconsistent reporting of crystal morphology in pharmaceutical samples [10] |
| Low Throughput [11] | Inability to rapidly process numerous samples creates a bottleneck in screening applications and limits statistical power. | - Manual axial scanning required for 3D models increases acquisition time and causes photobleaching [11]- Time-consuming focus-finding for each well in a multi-well plate [11] |
Troubleshooting Common Microscope Problems
This section addresses frequent issues encountered during experiments, their impact on research quality, and step-by-step resolutions.
Q1: My images are consistently blurry or out-of-focus, even after adjusting the knobs. What should I check?
- Problem: Blurry images undermine the accurate identification of protozoan cysts and oocysts, leading to potential misdiagnosis.
- Solution:
- Verify Sample Placement: Ensure the specimen is correctly positioned on the stage and the coverslip is properly seated [8].
- Clean Optics: Examine the eyepieces, objectives, and condenser for dust, fingerprints, or debris. Clean them meticulously with appropriate lens tissue and solution [8].
- Check for Misalignment: Misaligned optical components can cause distorted images. Follow the manufacturer's guidelines for aligning the condenser and objectives [8].
- Inspect the Sample: The issue may lie with the sample itself. Check for inadequate staining, uneven specimen thickness, or improper mounting [8].
Q2: I observe uneven illumination or a shadow in my field of view. How can I fix this?
- Problem: Uneven lighting reduces image quality and can interfere with quantitative analysis and automated image processing.
- Solution:
- Adjust the Condenser: Center the condenser and adjust its height according to Köhler illumination principles for optimal, even lighting [8].
- Check Diaphragm Settings: Ensure the field diaphragm is opened to just beyond the field of view.
- Inspect the Light Source: A faulty or aging bulb can cause uneven illumination. Replace the bulb if it shows signs of blackening or if it is near the end of its rated life [8].
Q3: My results are not reproducible between different operators. What steps can we take to standardize our workflow?
- Problem: This is a classic symptom of operator dependency and subjectivity, directly threatening the validity of your quality control data.
- Solution:
- Create Standard Operating Procedures (SOPs): Develop and enforce detailed SOPs for every step, from sample preparation and staining to microscopy settings and diagnostic criteria.
- Implement Cross-Training: Ensure all researchers are trained and calibrated against a set of gold-standard reference images.
- Utilize Automated Focus and Imaging: Where possible, use systems with autofocus and predefined imaging protocols to minimize human intervention [11].
Advanced Experimental Protocols to Overcome Limitations
Protocol: Metagenomic Next-Generation Sequencing (mNGS) for Parasite Detection
This protocol addresses the subjectivity and low throughput of traditional microscopic identification by using a universal, culture-independent test [9].
- Application: Simultaneous detection and differentiation of food and waterborne protozoan parasites (e.g., Cryptosporidium parvum, Giardia duodenalis, Toxoplasma gondii) on leafy greens for outbreak investigation.
- Principle: Metagenomic sequencing allows for comprehensive testing without prior knowledge of potential pathogens, overcoming the limitations of targeted molecular methods [9].
Workflow Overview:
Detailed Methodology [9]:
- Sample Preparation and Spiking: Romaine lettuce leaves (25 g) are spiked with a known number of parasite oocysts/cysts (e.g., 100-100,000 C. parvum oocysts) to establish detection limits.
- Washing and Concentration: Spiked leaves are washed in buffered peptone water with 0.1% Tween via stomacher. The filtrate is passed through a 35 μm filter and centrifuged at 15,000x g for 60 minutes to pellet the oocysts.
- Rapid Lysis: The pellet is lysed using the OmniLyse device for 3 minutes. This is a critical improvement over traditional freeze-thaw or heat lysis methods, which are time-consuming or can damage DNA.
- DNA Extraction and Amplification: DNA is extracted from the lysate via acetate precipitation. The extracted DNA is then subjected to whole genome amplification (WGA) to generate sufficient quantities (median 4.10 μg) for sequencing.
- Sequencing and Analysis: Amplified DNA is sequenced using MinION (Oxford Nanopore) or Ion S5 sequencers. The resulting fastq files are uploaded to the CosmosID bioinformatics platform for microbial identification and differentiation within the metagenome.
Protocol: Implementing Extended Depth of Field (EDOF) via PSF Engineering
This protocol tackles the low throughput associated with 3D imaging and manual focusing by enhancing the microscope's depth of field, allowing for single-snapshot imaging [11].
- Application: High-throughput imaging of 3D cellular models like tumor spheroids, where axial scanning is time-consuming and causes photobleaching.
- Principle: Point Spread Function (PSF) engineering modifies the image of a point source using a phase mask to encode 3D information, making the PSF robust to misfocus over an extended axial range [11].
Workflow Overview:
Detailed Methodology [11]:
- Compact PSF Engineering: A phase-modulating element (e.g., a photolithographically-fabricated phase mask) is placed in the back focal plane (BFP) at the exit of the infinity-corrected objective lens. This can be done using an annular mount threaded into the objective turret or a 3D-printed mask attached to the objective.
- Image Acquisition: Image the biological sample (e.g., fluorescently labeled nuclei in FaDu cell spheroids) using a single camera exposure without the need for axial (z-stack) scanning.
- Image Processing (Optional): For further improvement, apply post-processing algorithms like Lucy-Richardson deconvolution to the EDOF image to significantly enhance sharpness and recover 3D information.
The Scientist's Toolkit: Key Research Reagent Solutions
The table below lists essential materials for implementing the advanced protocols discussed.
| Item | Function / Application | Protocol |
|---|---|---|
| OmniLyse Device [9] | Rapid, efficient mechanical lysis of robust parasite oocyst/cyst walls within 3 minutes, enabling high-quality DNA extraction for sequencing. | Metagenomic NGS |
| Phase Mask [11] | An optical element placed in the objective's back focal plane to engineer the PSF, enabling extended depth-of-field or 3D snapshot imaging. | EDOF PSF Engineering |
| Whole Genome Amplification Kit | Amplifies tiny amounts of extracted DNA to the microgram quantities required for next-generation sequencing library preparation. | Metagenomic NGS |
| Nanopore Sequencer (MinION) [9] | A portable sequencing platform that generates metagenomic data within hours, allowing for real-time identification of pathogens. | Metagenomic NGS |
| Bioinformatics Platform (CosmosID) [9] | A highly curated software tool that analyzes metagenomic sequence data to identify and differentiate microbes at the genus, species, and genotype level. | Metagenomic NGS |
Frequently Asked Questions (FAQs)
Q: Can artificial intelligence (AI) really help overcome the subjectivity of traditional microscopy? A: Yes. Deep learning models, particularly convolutional neural networks (CNNs), are revolutionizing parasitic diagnostics by enhancing detection accuracy and consistency [2]. These models can be trained on large image datasets to automatically identify and characterize parasites, reducing bias and error from human judgment [10]. This is especially valuable for standardizing the analysis of morphological features, such as pharmaceutical crystal structures or parasite stages [10].
Q: What is the simplest way to improve throughput without buying a new, expensive system? A: Implementing an Extended Depth of Field (EDOF) PSF is a highly effective strategy [11]. By reducing or eliminating the need for time-consuming axial scanning and precise focus-finding on each sample, you can dramatically increase the speed of your imaging workflow. This can be achieved through a relatively simple modification to an existing microscope objective [11].
Q: Our lab relies on PCR for parasite identification. Why should we consider switching to mNGS? A: While PCR is highly sensitive for targeted detection, it requires prior knowledge of the organism and can typically only test for one or a few pathogens at a time [9]. mNGS is a universal, untargeted approach that can simultaneously identify and differentiate a wide range of parasites (and other microbes) in a single test, making it exceptionally powerful for outbreak investigations and surveillance studies where the causative agent is unknown [9].
Q: We keep getting fungus on our microscope lenses. How can we prevent this? A: Fungus growth is a serious issue caused by storing microscopes in damp or humid environments [8]. To prevent it, always store the instrument in a climate-controlled room with stable, low humidity. Using a microscope dehumidifier or storing objectives in a desiccator cabinet is highly recommended. If fungus appears, professional cleaning and maintenance are required [8].
Troubleshooting Guides and FAQs for Protozoan Identification
Sample Preparation and Handling
Q: My sample is contaminated with bacteria, overpowering the protozoans of interest. How can I resolve this?
A: Bacterial overgrowth is common in protozoan cultures like hay infusions. To troubleshoot:
- Preventive Culture Management: When creating hay infusions to culture protists, be aware that boiling the hay may not kill heat-resistant spores of bacteria like Bacillus and Clostridium. The bacterial biofilm that forms is a natural food source but should be contained [12].
- Aseptic Technique: Always use aseptic techniques to protect your sample from contamination and to protect yourself from potential infection. This includes using disinfectants like 70% ethanol on surfaces and hands, and avoiding the creation of aerosols [12].
- Controlled Disposal: Do not simply flush cultures down the drain. To safely dispose of liquid cultures, add chlorine bleach, allow it to act for several hours to ensure pathogen inactivation, and then dispose. Be aware that bleach is corrosive and requires careful handling [12].
Q: How can I verify that my fluorescent labeling is specific and not introducing artifacts?
A: Proper validation is crucial for quantitative microscopy.
- Use Unlabeled Controls: Always run unlabeled control samples (with no added fluorophore) to check for levels of autofluorescence from endogenous biological components [13].
- Validate Labeling Specificity: Do not assume antibodies or fluorescent proteins (FPs) are inert or perfectly specific. Nonspecific binding or perturbation of the target's natural function and localization is a common source of error [13].
- Test Sample Preparation Methods: The process of preparing and labeling the specimen itself can introduce errors. Validate your entire sample preparation protocol with known controls to ensure it accurately reveals the biology you are studying [13].
Imaging System Performance
Q: My quantitative microscopy data is inconsistent between sessions. What should I check?
A: Inconsistencies often stem from unvalidated imaging system performance.
- Establish a QC Schedule: Implement a regular quality control protocol for your microscope. This should include weekly visual inspections, monthly performance checks using control slides, and quarterly in-depth optical testing to verify illumination stability, alignment, and calibration [14].
- Identify Systematic Error: Microscopes are prone to systematic errors that can be hard to spot but significantly impact quantitative data. Use known samples to validate that your imaging system is capable of providing accurate measurements for your specific application [13].
- Document Everything: Maintain a log of all QC procedures, results, and any maintenance performed. This documentation is key to tracking system performance over time and identifying drift or issues [14].
Q: I am unsure if my microscope is correctly configured for detecting protozoan parasites. What are the key considerations?
A: The appropriate configuration depends on the target protozoan and the diagnostic goal.
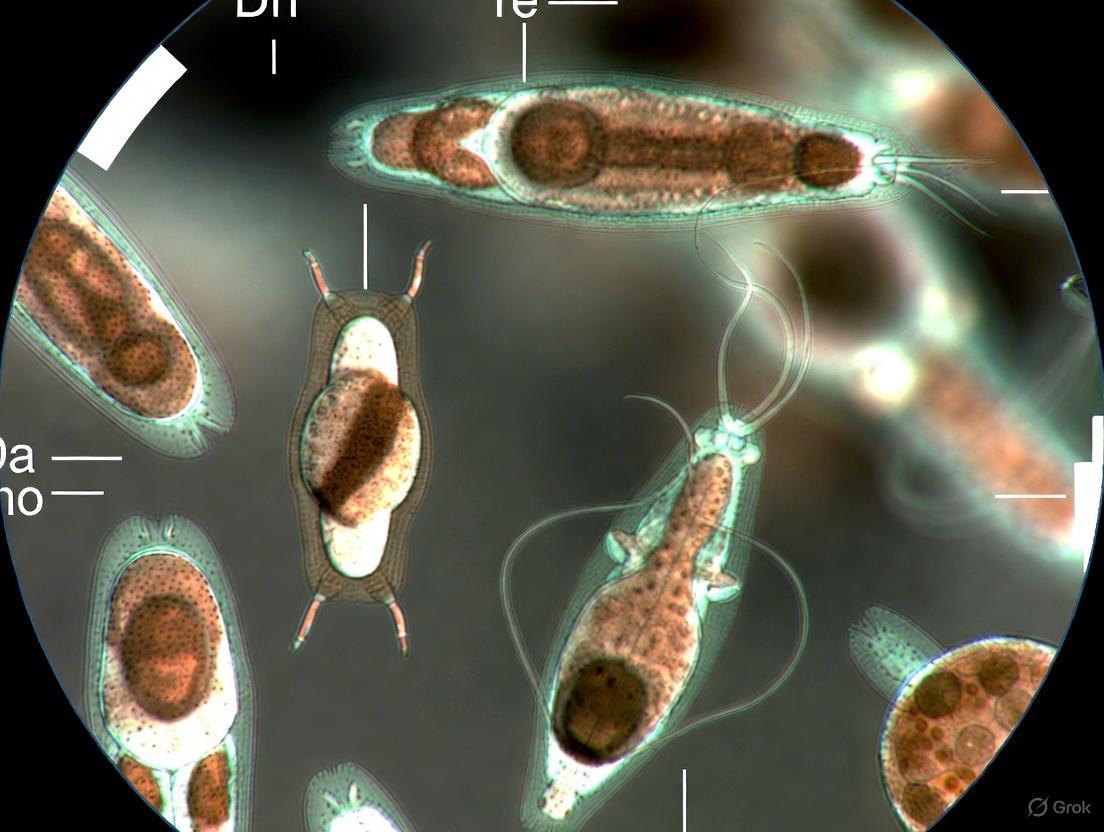
- Observe Live Specimens: For many flagellates and ciliates, in vivo observation is indispensable. Fixation can cause shrinkage and distortion, making important diagnostic characters unrecognizable. Characteristic motility is a key identifier that is best observed in fresh material [15].
- Select the Right Stain: Specific stains are required to visualize key structures.
- Protargol (silver protein) staining is essential for revealing the arrangements of flagella, cilia, and nuclei in flagellates and ciliates [15].
- Klein's silver nitrate stain is used for mobile peritrich ciliates to demonstrate the components of the adhesive disc [15].
- In tissue sections, special stains like Acid-fast for mature Cryptosporidium oocysts or Azure eosin/Giemsa for myxosporean polar capsules are used [15].
Diagnostic Methodology and Interpretation
Q: When should I choose molecular diagnostics over conventional microscopy for intestinal protozoa?
A: The selection depends on your objectives, required sensitivity, and resource availability. The table below compares the core diagnostic methods.
Table 1: Comparison of Diagnostic Methods for Common Intestinal Protozoa
| Diagnostic Method | Key Advantages | Key Limitations | Suitability for Entamoeba histolytica, Giardia duodenalis, Cryptosporidium |
|---|---|---|---|
| Conventional Microscopy [16] | Widely available; Low cost; Can detect a broad range of parasites. | Low sensitivity & specificity; Cannot differentiate pathogenic from non-pathogenic species (e.g., E. histolytica vs. E. dispar); Requires skilled examiner. | Low reliability for definitive species-level identification. |
| Immunodiagnostic (Antigen Detection) [16] | Higher sensitivity and specificity than microscopy; Faster than molecular methods; User-friendly rapid tests available. | May not differentiate between all species (e.g., some tests cannot distinguish E. histolytica from E. moshkovskii); May require fresh, unpreserved samples. | Good for specific detection of pathogens; Useful for intestinal amoebiasis and giardiasis. |
| Molecular Diagnosis (e.g., PCR) [2] [16] | Very high sensitivity and specificity; Can differentiate between morphologically identical species; Enables genotyping and epidemiological studies. | Higher cost; Requires specialized equipment and technical expertise; Not always point-of-care. | High reliability for definitive diagnosis and species identification. |
Q: How is modern technology like AI addressing the challenges of traditional microscopic diagnosis?
A: Artificial intelligence, particularly deep learning, is revolutionizing protozoan diagnostics by enhancing accuracy and efficiency.
- Automated Detection and Classification: Convolutional Neural Networks (CNNs) and object detection algorithms like YOLOv4 can automatically locate and classify protozoa in microscopic images with high accuracy (e.g., 97% in one study), reducing reliance on manual examination [17] [18].
- Handling Complex Images: These models can be trained to identify multiple protozoan species from freshwater, accounting for different shapes, sizes, and movements, even in images with varying light conditions or pollutants [17].
- Overcoming Subjectivity: AI models minimize the unconscious bias inherent in visual assessment of images, leading to more reproducible and objective results [13] [18].
Research Reagent Solutions for Protozoan Identification
The following table details essential reagents and their functions in protozoan research, spanning from classical to modern techniques.
Table 2: Key Research Reagents for Protozoan Identification and Analysis
| Reagent / Tool | Function / Application | Specific Example in Protozoology |
|---|---|---|
| Protargol (Silver Protein Stain) [15] | Stains basal bodies and infraciliary lattice of ciliates. | Essential for visualizing the arrangements of cilia, flagella, and nuclei in ciliates and flagellates for taxonomic identification [15]. |
| Klein's Silver Nitrate Stain [15] | Impregnates the adhesive disc of mobile peritrich ciliates. | Used to demonstrate the skeletal elements of the adhesive disc in trichodinids and other peritrichs [15]. |
| Bioorthogonal Non-Canonical Amino Acids (e.g., L-Aha, L-Anl) [19] | Incorporates chemical tags into newly synthesized proteins for tracking and enrichment. | BONCAT enables temporal tracking of the nascent proteome in parasites like Leishmania to study drug-induced adaptations [19]. |
| Proximity-Dependent Labeling Enzymes (e.g., BirA*) [19] | Biotinylates proteins in close proximity to a protein of interest. | BioID has been used in Toxoplasma gondii and Plasmodium to map the proteome of subcellular compartments like the parasitophorous vacuole membrane [19]. |
| Monoclonal Antibodies (for Immunodiagnostics) [16] | Targets specific parasite antigens in clinical samples. | Used in ELISA and rapid tests to detect E. histolytica Gal/GalNAc lectin in fecal specimens for diagnosis [16]. |
Experimental Workflow for Modern Protozoan Analysis
The following diagram illustrates a generalized integrated workflow for protozoan analysis, combining classical and modern technological approaches.
Key Internal and External Factors Complicating Protozoan Identification and Control
Troubleshooting Guide: Frequently Asked Questions
FAQ 1: Why is our laboratory's rate of protozoan identification inconsistent, even when analyzing the same specimen multiple times?
Several factors related to specimen handling and analyst skill can cause this inconsistency:
- Parasite Load and Shedding: Protozoa like Dientamoeba fragilis are irregularly shed. A single stool specimen examination detects only 58-72% of protozoal infections. Analyzing three specimens increases the yield significantly—by 31.1% for D. fragilis and 11.3% for Giardia [20].
- Specimen Dilution and Processing: The process of homogenizing and diluting specimens for testing can affect reproducibility. Studies show that low protozoal concentrations in a specimen are a major factor leading to poor concordance in repeat tests [21].
- Technologist Proficiency: Microscopy is subjective and requires a high level of skill. A shortage of experienced technologists, combined with infrequent encounters with positive specimens in non-endemic areas, hampers the ability to maintain proficiency. Internal quality control programs using blinded resubmission of clinical specimens have shown a benchmark concordance rate of about 80% for pathogenic protozoa under real-world conditions [21].
FAQ 2: What are the major limitations of traditional microscopy (Ova & Parasite examination) for protozoan diagnosis?
The O&P examination, while a cornerstone of diagnosis, faces significant challenges that can compromise result accuracy [20] [16]:
- Low and Variable Sensitivity: The sensitivity of the O&P examination is highly variable, reported to be between 20% and 90% when compared to more sensitive molecular assays [20].
- Inability to Differentiate Species: Microscopy cannot reliably distinguish between pathogenic and non-pathogenic species. For example, Entamoeba histolytica is morphologically identical to the non-pathogenic E. dispar without evidence of erythrophagocytosis [20] [16] [22].
- Labor-Intensive and Skill-Dependent: The procedure is slow, requires highly trained personnel, and is often deprioritized, leading to long turnaround times [20].
FAQ 3: Which common pathogenic protozoa are not detected by many rapid, FDA-cleared antigen tests, and how can we address this?
While antigen tests are excellent for Giardia, Cryptosporidium spp., and Entamoeba histolytica, a significant gap exists. There are no FDA-cleared antigen tests for Dientamoeba fragilis, a pathogenic protozoa frequently detected in many laboratories [20]. This necessitates reliance on the traditional O&P examination or the development and use of laboratory-developed molecular tests (e.g., PCR) to ensure this and other uncommon pathogens are not missed [20].
FAQ 4: How do environmental stressors complicate the study and control of protozoan parasites in aquatic ecosystems?
Research using mesocosm experiments shows that multiple environmental stressors interact in complex ways to affect protozoan communities [23]:
- Synergistic and Antagonistic Effects: The combination of warming and eutrophication has a synergistic effect, significantly promoting protozoan biomass. In contrast, the combination of warming and pesticide pollution has an antagonistic effect, reducing protozoan abundance, biomass, and diversity [23].
- Impact on Diversity and Function: Stressors like eutrophication, pesticides, and warming independently and interactively affect protozoan α-diversity, community structure, and the composition of functional groups (e.g., algivores, bacterivores) [23]. This makes predicting community trends under future climate scenarios difficult.
FAQ 5: Why is drug treatment for common mucosal protozoa becoming increasingly challenging?
Treatment is complicated by a limited arsenal of drugs and emerging resistance issues [24]:
- Reliance on a Single Drug Class: The nitroimidazole derivatives (especially metronidazole, or MTZ) are the most effective and widely used drugs for treating amebiasis, giardiasis, and trichomoniasis. For amebiasis, treatment is reliant on a single class of agents [24].
- Treatment Failure and Side Effects: There is emerging evidence of an increased frequency of therapeutic failure with MTZ. Furthermore, adverse side effects and problems with use during pregnancy are common concerns [24].
- Limited Options: For some parasites, like Cryptosporidium spp., effective treatment options are extremely limited, with nitazoxanide being one of the few drugs available [24].
Table 1: Sensitivity of Conventional Diagnostic Methods for Key Intestinal Protozoa
| Organism | Common Diagnostic Method | Reported Sensitivity | Key Diagnostic Limitation |
|---|---|---|---|
| Entamoeba histolytica | Microscopy (O&P) | N/A | Cannot differentiate from non-pathogenic E. dispar and E. moshkovskii [16] |
| Giardia duodenalis | Permanent stained smear (Chlorazol black dye) | 66.4% [16] | Sensitivity is highly dependent on stain quality and examiner skill. |
| Cryptosporidium spp. | Modified acid-fast stain | 54.8% [16] | Small, poorly stained oocysts are easily missed; requires special stain request. |
| Multiple Pathogens | Single stool specimen for O&P | 58-72% [20] | Detects only a fraction of true infections due to irregular shedding. |
Table 2: Internal Quality Control (QC) Concordance for Pathogenic Protozoa (Blinded Resubmission Study)
| Targeted Protozoan | Concordance Rate in QC Program | Major Factor Affecting Concordance |
|---|---|---|
| Entamoeba histolytica/E. dispar | ~80% [21] | Low protozoal concentration in the specimen [21] |
| Giardia lamblia | ~80% [21] | Low protozoal concentration in the specimen [21] |
| Dientamoeba fragilis | ~80% [21] | Low protozoal concentration in the specimen [21] |
Experimental Protocol: Intra-Laboratory Quality Control Using Blinded Resubmission
This protocol assesses the reproducibility of microscopic identification and can be integrated into a laboratory's quality assurance program [21].
1. Specimen Selection and Storage:
- Select clinical stool specimens preserved in Sodium-Acetate-Acetic acid-Formalin (SAF). SAF-preserved specimens can be stored for several months for this purpose.
- Create a balanced collection, including both positive and negative specimens for various protozoa, with a range of parasite concentrations.
2. Creation of Blinded Test Subsets:
- Resubmitted Pair: Dilute the original SAF-preserved specimen with a small volume of fresh SAF to ensure homogeneity and create a new pair of specimens. Relabel these with new, fictional accession numbers and patient information.
- Pooled Specimen: For a pair of specimens from the same patient, pool equal amounts from each to create a single, pooled specimen. Relabel this as above.
3. Integration into Workflow:
- Introduce the blinded resubmission and pooled specimens into the routine laboratory workflow, ensuring they are processed and examined by technologists who are unaware of their status as quality control samples.
4. Data Analysis and Concordance Calculation:
- Compare the results of the initial report with the report from the blinded resubmission.
- A set is considered concordant if the results (positive/negative and identification) of both the initial and resubmitted reports are the same.
- Calculate the concordance rate as the percentage of concordant sets out of all sets positive for a particular parasite. A benchmark of ~80% concordance for key pathogens can be used as a target [21].
Workflow Diagram
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Protozoan Identification and Research
| Reagent / Material | Primary Function | Example Application in Protozoology |
|---|---|---|
| SAF (Sodium Acetate-Acetic Acid-Formalin) Preservative | Long-term preservation of stool specimens for morphology. | Preferred preservative for storing clinical specimens for blinded quality control resubmission programs [21]. |
| Iron-Hematoxylin & Trichrome Stain | Permanent staining for detailed nuclear and cellular morphology. | Used for permanent stained slides critical for identifying internal structures of protozoa like Dientamoeba fragilis [22] [21]. |
| Modified Acid-Fast Stain | Selective staining of oocyst walls of coccidian parasites. | Differentiates Cryptosporidium spp., Cyclospora cayetanensis, and Isospora belli oocysts, which appear bright red [22]. |
| Monoclonal Antibodies (e.g., vs. Gal/GalNAc lectin) | Target-specific detection of parasite antigens in immunoassays. | Used in ELISA and rapid immunochromatographic tests to detect Entamoeba histolytica specifically, distinguishing it from E. dispar [16]. |
| Protargol (Silver Protein) Stain | Stains basal bodies and ciliary/flagellar structures. | Essential for visualizing the infraciliary lattice of ciliates and the arrangement of flagella in flagellates for precise species identification [25]. |
Implementing Advanced Methodologies: Digital Workflows and AI in the Modern Lab
This technical support center provides troubleshooting and best practices for researchers in the microscopic identification of protozoans, ensuring the quality and reproducibility of your digital pathology data.
Troubleshooting Guides
Common Slide Scanning Issues and Solutions
| Problem Area | Specific Issue | Possible Cause | Solution |
|---|---|---|---|
| Image Focus | Entire slide or large areas are out of focus [26] | Slide not sitting flat in scanner; standard tissue thickness (3–5 μm) exceeded for single-plane scanning [26] | Ensure slide is flush in scanner rack; for thick sections, use multi-plane (Z-stack) scanning [26] |
| Image Focus | Specific tissue areas are blurry [26] | Tissue folds, air bubbles under coverslip, or debris on slide surface [26] | Manually place additional focus points on flat tissue areas; avoid points on debris or defects [26] |
| Tissue Detection | Scanner fails to automatically detect tissue regions [26] | Faint staining, excessive background stain, or debris/marks on slide confusing the algorithm [26] | Review tissue detection preview on suboptimal slides prior to full-resolution scanning [26] |
| Image Artifacts | "Streaking" artifacts in oil scanning [26] | Objective drying out during the scanning process due to insufficient oil [26] | Apply enough oil before scanning to prevent drying; perform test scans [26] |
| Image Artifacts | "Glazed" appearance with poor contrast [26] | Too much oil, which can seep under the coverslip [26] | Use less oil; clean oil residue from scanner racks after use [26] |
| Image Artifacts | "Stitch lines" in the final image [26] | Misalignment of the scanned stripes that make up the full image [26] | This is often a scanner hardware/software issue; ensure the scanner is calibrated [26] |
| Slide Handling | Risk of damage to scanner or slide [26] | Cracked/chipped slides, overhanging labels, or tape impeding the mechanism [26] | Inspect slides for damage and remove any loose glass, tape, or overhanging labels before scanning [26] |
Workflow for Quality Control of Scanned Protozoan Slides
This workflow provides a standardized method for verifying the quality of your digital slides, which is critical for accurate protozoan identification.
Frequently Asked Questions (FAQs)
Pre-Scanning Preparation
Q: What are the most critical pre-scanning steps to ensure a high-quality digital slide of protozoan samples? A: The most critical steps involve sample and slide preparation [26]:
- Staining: Ensure staining is neither too faint nor has excessive background, as this hinders automatic tissue detection. This is crucial for visualizing delicate protozoan structures.
- Coverslipping: Use glass coverslips, as plastic can warp. Ensure no air bubbles are trapped and that the mounting medium is fully dry before scanning.
- Slide Inspection: Clean slides with a soft cloth to remove debris or fingerprints. Check for cracks or overhanging labels that could damage the scanner.
Q: How does sample thickness affect the scanning of protozoans? A: Scanners have a smaller depth of focus than traditional microscopes [26]. For standard single-plane scanning, 3–5 μm sections are ideal. If your sample preparation results in thicker sections, you must use a scanner capable of multi-plane (Z-stack) scanning to capture all structures in focus.
Scanning Process
Q: The scanner is not automatically detecting all the protozoan cysts on my slide. What should I do? A: This is common with faintly stained samples or those with debris [26]. Manually review the tissue detection map the scanner generates prior to the high-resolution scan. You can often adjust the detection area manually to ensure all relevant sections are included.
Q: What is the best way to ensure optimal focus across the entire sample? A: While automatic focusing is standard, you can improve results by [26]:
- Ensuring the slide is perfectly flat during scanning.
- Manually placing additional focus points evenly across the tissue.
- Avoiding placing focus points on areas with debris, air bubbles, or tears.
Quality Control and Traceability
Q: What is the minimum QC check I should perform on a scanned slide before analysis? A: At a minimum [26]:
- Review the entire slide at low magnification (e.g., 4x) for obvious focus issues or misalignment ("stitch lines").
- Zoom to the capture magnification and pan horizontally and vertically across the widest part of the sample, checking for smaller out-of-focus areas.
- Perform high-magnification spot checks on regions of interest, such as where the tissue thickness varies.
Q: How does digital slide scanning contribute to a traceable foundation in research? A: Digital scanning creates a permanent, unalterable record of your slide at a specific point in time [26]. This supports traceability and standardization by:
- Allowing exact same sample to be reviewed by multiple researchers.
- Enabling re-evaluation of findings years later without degradation.
- Facilitating the sharing of identical image data for collaboration or publication, ensuring all parties are analyzing the same source material.
The Scientist's Toolkit: Research Reagent & Material Solutions
Essential Materials for Protozoan Sample Preparation and Scanning
| Item | Function & Importance |
|---|---|
| Glass Coverslips | Essential for creating a flat scanning plane. Plastic coverslips can warp over time, leading to focus issues, and should be avoided for permanent digital records [26]. |
| High-Quality Mounting Medium | Preserves the sample under the coverslip. Must be fully dry before scanning to avoid leaving residue on the scanner mechanism [26]. |
| Immersion Oil | Required for high-magnification (e.g., 100x) scanning to achieve optimal resolution. Both under- and over-application can cause artifacts, so test scans are recommended [26]. |
| Soft Lint-Free Cloths | Used for cleaning slides before they enter the scanner. Removes dust, water spots, and fingerprints from both the top and bottom surfaces, which can obscure image quality [26]. |
| Standardized Staining Kits | Using consistent, high-quality stains (e.g., Trichrome for protozoans) is vital. Variable or faint staining directly impacts the scanner's ability to detect tissue and compromises digital analysis [26] [27]. |
Workflow for Digital Slide Creation in Protozoan Research
This diagram outlines the key stages in creating a traceable digital slide, from sample to digital asset.
Convolutional Neural Networks (CNNs) for Automated Detection and Classification
Frequently Asked Questions (FAQs)
Q1: My CNN's loss value is not improving during training. What are the first things I should check? If your loss value is not improving, start with these fundamental checks [28]:
- Verify the Loss Function and Optimizer: Ensure you are using an appropriate loss function (e.g., cross-entropy for classification) and a modern optimizer (e.g., Adam, SGD with momentum).
- Check Learning Rate: A too-high learning rate can cause the loss to oscillate or diverge, while a too-low rate leads to minimal change. Adjust the initial learning rate and implement a learning rate decay schedule.
- Confirm Variable Training: Use tools like TensorBoard to check that all trainable variables are updating. If not, check if they are correctly registered as trainable and check for vanishing gradients.
- Overfitting Check: If the training loss decreases but validation loss increases, you are overfitting. See the troubleshooting guide on overfitting for solutions.
Q2: What does it mean if my model is overfitting, and how can I prevent it? Overfitting occurs when your model "memorizes" the training data but fails to generalize to new data, typically indicated by a growing gap between training and validation accuracy [28]. To prevent it:
- Implement Data Augmentation: Apply random transformations (e.g., rotation, flipping, cropping) to your training images to increase data diversity.
- Use Regularization Techniques: Add Dropout layers to randomly ignore neurons during training, or use L2 regularization in your layers.
- Apply Batch Normalization: This technique can improve stability and performance.
- Employ Early Stopping: Halt training when the validation performance stops improving.
Q3: My CNN fails to correctly localize objects in a simple coordinate transformation task. Is this a known issue? Yes, this is a known limitation of standard CNNs. A study from Uber AI Labs demonstrated that CNNs can fail spectacularly on a seemingly simple task of mapping between (x,y) coordinates and one-hot pixel space [29]. The solution is to use a CoordConv layer, which adds extra input channels carrying spatial coordinate information (e.g., i and j coordinates), allowing the network to learn translation variance when needed. This fix led to perfect generalization with far fewer parameters and faster training times [29] [30].
Troubleshooting Guides
Issue 1: Model Not Converging (Loss Not Decreasing)
Follow this systematic workflow to diagnose and resolve convergence issues:
Protocols and Detailed Methodologies:
- Check Data and Label Pairing: Manually inspect a small batch of your input data and corresponding labels to ensure they are correctly matched and preprocessed. A common error is shuffled labels or incorrect normalization [28].
- Overfit a Small Dataset:
- Purpose: To verify the model's capacity and the integrity of the training pipeline.
- Procedure: Turn off regularization/dropout. Take a very small portion of your training set (e.g., 5-10 samples) and train for multiple epochs. You should be able to drive the loss close to zero or achieve 100% accuracy on this tiny set. If not, there is a fundamental issue with your model or data [28].
- Perform Gradient Check:
- Purpose: To ensure that your backpropagation is correctly computing gradients, especially when using custom operations.
- Procedure: Compare the gradients computed by your backpropagation algorithm against numerically approximated gradients using the formula:
[grad f(x)]_i ≈ (f(x+eps*e_i) - f(x-eps*e_i)) / (2*eps). Significant discrepancies indicate a bug in your gradient calculation [31].
- Tune Hyperparameters: The optimal learning rate is often close to the largest rate that does not cause training divergence. Start with a large rate and reduce it by a factor if divergence occurs until stable training is achieved [28].
Issue 2: Vanishing or Exploding Gradients
This problem is characterized by upstream network weights (closer to the input) changing very slowly or becoming excessively large during training, hindering learning [28].
Solutions:
- Improved Weight Initialization: Use established initialization methods (e.g., He, Xavier) instead of initializing all weights to zero or the same value.
- Alternative Activation Functions: Replace sigmoid or tanh functions with ReLU, Leaky ReLU, or MaxOut activations, which have better gradient propagation properties.
- Architectural Changes: Incorporate Batch Normalization layers to stabilize and normalize the inputs to subsequent layers. For recurrent networks, use LSTM blocks [28].
Issue 3: Specific Challenges in Protozoan Image Detection
Microscopic images of protozoa present unique challenges, including varying light conditions, deformation of organisms, and contaminants in the water, which can affect model performance [17]. The table below summarizes key performance metrics from a recent study on protozoa detection for benchmarking purposes.
Table: Performance Metrics of a YOLOv4 Model for Protozoa Detection [17]
| Metric | Score | Description |
|---|---|---|
| Accuracy | 97% | Overall correctness of the model. |
| mAP (mean Average Precision) | 0.9752 | Overall detection accuracy across all classes. |
| Precision | 0.92 | Proportion of correct positive identifications. |
| Sensitivity (Recall) | 0.98 | Proportion of actual positives correctly identified. |
| F1-Score | 0.95 | Harmonic mean of precision and sensitivity. |
Experimental Protocols for Quality Control
This section outlines a reproducibility assessment protocol adapted from clinical parasitology, which can be integrated into deep learning research for robust model validation.
Protocol: Blinded Resubmission for Reproducibility Assessment
1. Objective: To evaluate the consistency and reproducibility of your CNN model's detections by testing it on blinded, resubmitted samples from your dataset [21].
2. Materials:
- A curated dataset of protozoan images.
- Your trained CNN detection model.
- Data processing and augmentation pipeline.
3. Methodology:
- Sample Selection: From your main dataset, select a subset of images. Balance this subset between positive and negative samples, and include a range of protozoan concentrations if possible [21].
- Blinded Resubmission: After the initial model testing, the selected images are subtly modified. This can include:
- Dilution: Adding noise or slightly altering contrast to simulate lower "protozoal concentration" [21].
- Pooling: Creating new test images by combining parts of two different original images to check if the model can correctly identify all entities [21].
- These modified images are then relabeled with new accession numbers and mixed back into the evaluation queue as if they were new samples.
- Concordance Analysis: Compare the model's reports from the initial evaluation and the blinded resubmission. Concordance is defined as the percentage of samples where the results (positive/negative and classification) are identical. A benchmark concordance rate of around 80% for pathogenic protozoa has been suggested in clinical studies and can serve as an initial goal [21].
The workflow for this quality control protocol is as follows:
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Materials for Protozoan Detection Experiments
| Item | Function / Explanation |
|---|---|
| SAF Preservative | Sodium acetate-acetic acid-formalin; used for long-term preservation of clinical stool specimens for parasitology analysis, allowing storage for several months [21]. |
| Iron-Hematoxylin Stain | A permanent staining technique used to enhance the contrast and visibility of protozoal structures during microscopic examination [21]. |
| 18S Amplicon NGS Assay | A metabarcoding approach for the simultaneous detection of multiple protozoan pathogens (e.g., Cryptosporidium, Giardia, Toxoplasma gondii) in a single sample, using next-generation sequencing [32]. |
| YOLOv4 Algorithm | A state-of-the-art deep learning object detection model known for its exceptional speed and accuracy, suitable for real-time detection of protozoa from microscopic images [17]. |
| CoordConv Layer | A modified convolutional layer that provides the model with access to its own input coordinates, solving fundamental failures of standard CNNs in certain spatial tasks [29] [30]. |
| Data Augmentation Pipeline | A software toolset for applying random transformations (mirroring, rotation, cropping, elastic deformation) to training images, which is critical for improving model generalization and preventing overfitting [28] [17]. |
Troubleshooting Guides
Low DNA Yield from Fecal Samples
Problem: Inadequate quantity or quality of DNA extracted from fecal samples for molecular detection of protozoa.
Solutions:
- Verify Preservation Method: Ensure samples are preserved in an appropriate buffer. A 2024 study directly compared preservation media and found that lysis buffer was superior to 99.8% ethanol, yielding DNA concentrations up to three times higher and with better integrity for downstream PCR and sequencing [33].
- Optimize Lysis for Robust Cysts/Oocysts: The rigid wall of protozoan cysts and oocysts can resist standard lysis methods. A 2025 study on foodborne protozoa demonstrated that using a dedicated physical lysis device (OmniLyse) rapidly and efficiently broke down these walls, leading to a more sensitive detection of parasites like Cryptosporidium and Giardia [9].
- Use an Internal Extraction Control: Incorporate an internal control during the DNA extraction process to distinguish true PCR negatives from failures caused by inhibition or inefficient DNA recovery [34].
Discrepancies Between Microscopy and Molecular Results
Problem: Microscopy and PCR results for the same sample do not match.
Solutions:
- Understand Methodological Strengths: Recognize that each technique has different sensitivities. A large 2025 prospective study found multiplex PCR detected significantly more protozoan infections (Blastocystis spp.: 19.25%; Dientamoeba fragilis: 8.86%) compared to microscopy (6.55% and 0.63%, respectively) [35].
- Check PCR Panel Targets: Confirm the multiplex PCR panel used includes all protozoa of interest. The same study noted that microscopy was essential for detecting parasites not targeted by the PCR panel, such as Cystoisospora belli and helminths [35].
- Confirm Specificity for Debated Protozoa: For protozoa like Dientamoeba fragilis and Blastocystis spp., consider confirming positive PCR results with an alternative, specific simplex qPCR to rule out false positives [35].
Poor Contrast in Transmission Electron Microscopy (TEM)
Problem: Inadequate visualization of ultrastructural details in parasitic protozoa like Giardia intestinalis and Trichomonas vaginalis.
Solutions:
- Use Tannic Acid as a Mordant: Incorporate 1% tannic acid into the primary glutaraldehyde fixative. A 2025 protocol demonstrated this significantly enhanced in-block contrast of plasma membranes, organelle boundaries, and cytoskeletal elements without introducing artifacts [36].
- Simplify Staining Protocols: The contrast enhancement from tannic acid can be so effective that it sometimes allows for the omission of subsequent toxic stains like lead citrate, streamlining the workflow [36].
- Apply as a Post-Staining Agent: Tannic acid can also be used as a replacement for uranyl acetate for staining ultrathin sections, maintaining high image quality while avoiding radioactive reagents [36].
Frequently Asked Questions (FAQs)
Q1: Should I choose a commercial multiplex PCR or an in-house PCR assay for diagnosing intestinal protozoa?
A1: Both have their place, and the choice depends on your laboratory's resources and needs. A 2025 multicentre comparison found that a commercial test (AusDiagnostics) and a validated in-house RT-PCR showed complete agreement for detecting Giardia duodenalis [34]. Commercial kits offer standardization and ease of use, while in-house assays provide flexibility but require extensive validation and may show variable performance, especially for parasites like D. fragilis where DNA extraction efficiency is critical [34].
Q2: What is the most effective way to preserve stool samples for molecular analysis of the protozoan microbiome?
A2: For studies focusing on microbial community profiles, including protozoa, preservation in a lysis buffer is highly recommended over ethanol. Research from 2024 showed that lysis buffer not only provided higher DNA yield and quality but also better preserved the microbial community structure for accurate 16S and 18S rRNA sequencing [33].
Q3: In the era of molecular diagnostics, is microscopic examination still necessary?
A3: Yes, microscopy remains a crucial complementary technique. While molecular methods like multiplex PCR are more sensitive for detecting specific protozoa, microscopy is indispensable for identifying parasites not included in PCR panels (e.g., Cystoisospora belli, non-pathogenic protozoa, and helminths) [35]. It is particularly important for specific patient groups, such as those who are HIV-infected or returning travelers [35].
Q4: Are there emerging technologies that can automate protozoa detection?
A4: Yes, deep learning and metagenomic sequencing are promising technologies.
- Deep Learning: A 2024 study applied the YOLOv4 algorithm to automatically detect and classify freshwater protozoa from microscopic images with 97% accuracy, offering a tool for rapid, high-throughput analysis [17].
- Metagenomic Next-Generation Sequencing (mNGS): This culture-independent method can identify and differentiate multiple protozoan parasites from a single sample without prior knowledge of the target. A 2025 study successfully used nanopore sequencing to detect as few as 100 Cryptosporidium oocysts on lettuce, functioning as a universal detection test [9].
Table 1: Detection Rates of Intestinal Protozoa by Multiplex PCR vs. Microscopy (n=3,495 samples) [35]
| Protozoan | Multiplex PCR Detection Rate | Microscopy Detection Rate |
|---|---|---|
| Blastocystis spp. | 19.25% | 6.55% |
| Dientamoeba fragilis | 8.86% | 0.63% |
| Giardia intestinalis | 1.28% | 0.7% |
| Cryptosporidium spp. | 0.85% | 0.23% |
| Entamoeba histolytica | 0.25% | 0.68%* |
*Microscopy cannot differentiate *E. histolytica from non-pathogenic E. dispar [35].*
Table 2: DNA Yield and Quality from Fecal Samples Preserved in Different Media [33]
| Metric | Lysis Buffer | 99.8% Ethanol |
|---|---|---|
| DNA Concentration | Significantly higher | Lower (up to 3x difference) |
| DNA Integrity | Superior | Lower |
| A260/280 Purity | Optimal (Mean: 1.92, SD: 0.27) | Good but variable (Mean: 1.94, SD: 1.10) |
| 16S/18S PCR Success | Higher number of positive reactions | Fewer positive reactions |
Detailed Experimental Protocols
This protocol enhances contrast for ultrastructural analysis of protozoa like Giardia and Trichomonas.
Key Research Reagent Solutions:
- Primary Fixative: 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2) supplemented with 1% tannic acid.
- Post-fixative: 1% osmium tetroxide (OsO₄) and 0.8% potassium ferricyanide in 0.1 M cacodylate buffer.
- Dehydration Series: Gradual acetone solutions (e.g., 50%, 70%, 90%, 100%).
- Embedding Media: Epoxy resin.
Methodology:
- Fixation: Wash cell pellets in PBS and fix in the primary fixative (with tannic acid) for 2 hours at room temperature.
- Washing: Rinse cells several times with 0.1 M cacodylate buffer.
- Post-fixation: Treat cells with the post-fixative solution for 30 minutes.
- Dehydration: Dehydrate the sample through a graded series of acetone.
- Infiltration and Embedding: Infiltrate cells with epoxy resin and polymerize at 60°C for 72 hours.
- Sectioning and Staining: Cut ultrathin sections. Staining with uranyl acetate and/or lead citrate may be omitted or shortened based on the contrast achieved.
This protocol describes a sensitive mNGS method for detecting foodborne protozoa.
Key Research Reagent Solutions:
- Parasite Suspension: Purified oocysts/cysts (e.g., Cryptosporidium, Giardia) in phosphate-buffered saline (PBS).
- Wash Buffer: Buffered peptone water with 0.1% Tween.
- Lysis Device: OmniLyse for rapid mechanical lysis.
- Whole Genome Amplification Kit: To generate sufficient DNA for sequencing.
Methodology:
- Sample Spiking: Inoculate the surface of 25g lettuce leaves with a known number of parasite oocysts/cysts and air-dry.
- Elution and Concentration: Place lettuce in a stomacher bag with wash buffer and homogenize. Filter the fluid to remove debris and pellet microbes by high-speed centrifugation.
- Rapid Lysis: Lyse the pellet using the OmniLyse device for 3 minutes.
- DNA Extraction and Amplification: Extract DNA via acetate precipitation. Subject the extracted DNA to whole genome amplification.
- Sequencing and Analysis: Prepare libraries and sequence using a platform like MinION (Oxford Nanopore). Analyze raw reads using a bioinformatics platform (e.g., CosmosID) to identify parasites in the metagenome.
Workflow Visualization
Diagram 1: Integrated workflow for protozoan identification and QC.
Research Reagent Solutions
Table 3: Essential Reagents for Protozoan Identification Techniques
| Reagent | Function | Application Context |
|---|---|---|
| Lysis Buffer | Preserves DNA and facilitates cell lysis in fecal samples. Superior to ethanol for molecular studies [33]. | Molecular Diagnostics (PCR, NGS) |
| Multiplex PCR Panel | Simultaneously detects multiple protozoan DNA targets from a single sample [35]. | Molecular Diagnostics |
| Tannic Acid | Mordant that enhances contrast of membranes and cytoskeleton in TEM samples [36]. | Advanced Imaging (TEM) |
| OmniLyse Device | Provides rapid mechanical lysis of robust protozoan oocysts/cysts for efficient DNA release [9]. | Sample Preparation for NGS |
| Formalin-Ethyl Acetate (FEA) | Solution used for concentration and preservation of parasitic forms for microscopic examination [34]. | Traditional Microscopy |
In the context of research focused on the quality control of microscopic identification of protozoans, molecular techniques serve as powerful complementary tools. While microscopy provides a foundational morphological assessment, Polymerase Chain Reaction (PCR) and metagenomic next-generation sequencing (mNGS) offer unparalleled specificity, sensitivity, and the capacity for high-throughput analysis. This technical support center addresses common experimental challenges encountered when integrating these molecular methods into a protozoan research workflow, providing targeted troubleshooting guides and FAQs to ensure data accuracy and reliability.
PCR Troubleshooting Guide
Frequently Asked Questions (FAQ)
Q1: Why is there no PCR product or a very low yield on my gel? A1: This common issue can stem from several sources [37] [38]. First, confirm the integrity and purity of your DNA template using spectrophotometry (a 260/280 ratio of ~1.8 is ideal) or gel electrophoresis [39]. Ensure no PCR inhibitors, such as phenol or salts, are present. Next, optimize your reaction conditions: increase the number of cycles (e.g., to 35-40), check that all reaction components were added, and use a sufficient amount of DNA template [39]. Verify your primer design and concentration, and optimize the annealing temperature, often 3–5°C below the primer's Tm [39].
Q2: My PCR results show multiple bands or smearing. How can I improve specificity? A2: Non-specific products and smearing often indicate low reaction stringency [37]. To resolve this, incrementally increase the annealing temperature [39]. Switch to a hot-start DNA polymerase to prevent primer-dimer formation and non-specific amplification at low temperatures [39] [37]. Ensure your primer design is optimal, with minimal self-complementarity, and avoid high primer concentrations [39]. If smearing is persistent and was not an issue before, it may be due to accumulated amplifiable contaminants in the lab environment; consider using a new set of primers with different sequences [37].
Q3: What can I do to amplify a difficult, GC-rich protozoan gene target? A3: GC-rich regions and sequences with secondary structures are challenging [39]. Utilize DNA polymerases with high processivity, which have a stronger affinity for complex templates. Incorporate PCR additives or co-solvents such as DMSO (1-10%), formamide (1.25-10%), or betaine (0.5 M to 2.5 M) to help denature stable secondary structures [39] [40]. Increase the denaturation temperature and/or time to ensure complete separation of the DNA strands [39].
Troubleshooting Table: Common PCR Problems and Solutions
The following table summarizes quantitative data and recommendations for resolving frequent PCR issues.
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| No/Low Yield [37] [38] | Insufficient template | Increase input DNA to 1–1000 ng [39]. For genomic DNA, use 1 ng–1 µg per 50 µL reaction [38]. |
| Suboptimal cycling | Increase cycles to 25-40; ensure annealing temp is 3-5°C below primer Tm [39]. | |
| Enzyme inhibition | Re-purify DNA to remove contaminants (phenol, EDTA); use inhibitors-tolerant polymerases [39]. | |
| Non-Specific Bands/Smearing [39] [37] | Low annealing temperature | Increase temperature in 1-2°C increments; use a gradient cycler [39]. |
| Excess enzyme/Mg²⁺ | Decrease amount of DNA polymerase; optimize Mg²⁺ concentration (e.g., 0.2-5.0 mM) [39] [38]. | |
| Poor primer design | Redesign primers to avoid secondary structures and ensure specificity to target [39] [40]. | |
| Primer-Dimer Formation [39] [37] | High primer concentration | Optimize primer concentration, typically between 0.1–1 µM [39]. |
| Low annealing temperature | Increase annealing temperature to improve specificity [39]. | |
| Long annealing time | Shorten the annealing time to minimize non-specific binding [39]. |
Essential Research Reagent Solutions for PCR
The following reagents are critical for successful PCR experiments in protozoan research.
| Reagent | Function & Importance | Optimization Tips |
|---|---|---|
| DNA Polymerase | Enzyme that synthesizes new DNA strands. | Choose hot-start for specificity; high-processivity for difficult (GC-rich) templates; high-fidelity for cloning [39]. |
| Mg²⁺ Ions | Essential cofactor for DNA polymerase activity. | Concentration is critical; optimize between 0.5-5.0 mM. Excess can cause non-specificity [39] [40]. |
| PCR Additives | Co-solvents that modify DNA melting behavior. | Use DMSO, betaine, or formamide to denature GC-rich regions and secondary structures [39] [40]. |
| dNTPs | Building blocks (nucleotides) for new DNA strands. | Use balanced equimolar concentrations (200 µM of each dNTP total) to prevent incorporation errors [39] [40]. |
PCR Experimental Workflow
The diagram below outlines the key steps and decision points in a standard PCR experiment, from setup to analysis.
Metagenomic Sequencing Troubleshooting Guide
Frequently Asked Questions (FAQ)
Q1: My NGS library yield is unexpectedly low. What are the main causes? A1: Low library yield is a frequent issue in sequencing preparation [41]. The primary causes include:
- Poor Input DNA Quality: Degraded DNA or contaminants like salts or phenol can inhibit enzymes during library preparation. Re-purify your sample and check purity ratios (260/230 > 1.8) [41].
- Inefficient Fragmentation & Ligation: Over- or under-fragmentation reduces ligation efficiency. Optimize your fragmentation parameters and verify the adapter-to-insert molar ratio to avoid excessive adapter dimers [41].
- Overly Aggressive Cleanup: Size selection and purification steps can lead to significant sample loss. Ensure you are using the correct bead-to-sample ratio and avoiding over-drying of beads [41].
Q2: How does my choice of reference database affect metagenomic classification for protozoans? A2: The reference database is your ground truth and profoundly impacts results [42]. Common database issues include:
- Taxonomic Mislabeling: An estimated 1-3.6% of sequences in public databases may be misannotated, leading to false positives [42] [43]. For example, an E. coli genome might be misidentified, affecting downstream analysis.
- Database Contamination: Sequences from host DNA, vectors, or other organisms can be present within database entries, causing false detections. One study found over 2 million contaminated sequences in GenBank [42].
- Taxonomic Underrepresentation: Many non-model or rare protozoans may be absent or poorly represented, leading to false negatives. Mitigate this by using curated databases or supplementing with targeted sequencing [42].
Q3: What is a cost-effective sequencing strategy for detecting protozoan pathogens in clinical samples? A3: While increasing read length and data volume generally improves detection, it also increases cost and analysis time. A recent study on bronchoalveolar lavage fluid samples found that a strategy of 20 million reads in single-end 75 bp (SE75) mode provided a excellent balance, achieving high recall rates while remaining cost-effective [44]. The study also noted that samples with high pathogen nucleic acid loads were less affected by sequencing strategy choices [44].
Troubleshooting Table: Common mNGS Preparation Issues
| Problem | Failure Signals | Common Root Causes & Corrective Actions |
|---|---|---|
| Low Library Yield [41] | Low molarity; faint/broad electropherogram peaks. | Cause: Enzyme inhibition from contaminants.Fix: Re-purify input DNA; use fluorometric quantification (Qubit). |
| Cause: Inefficient ligation or tagmentation.Fix: Titrate adapter:insert ratio; optimize enzyme conditions. | ||
| Adapter Dimers/Contamination [41] | Sharp peak at ~70-90 bp in electropherogram. | Cause: Excess adapters or inefficient cleanup.Fix: Optimize bead-based cleanup ratios; use double-size selection. |
| High Duplicate Rate/Bias [41] | Overamplification artifacts; skewed sequence distribution. | Cause: Too many PCR cycles during library amplification.Fix: Reduce the number of amplification cycles; use high-fidelity polymerases. |
| Poor Taxonomic Classification [42] [43] | False positives/negatives; strange cross-kingdom assignments. | Cause: Use of an uncurated database with mislabeled sequences.Fix: Use curated databases; apply tools like GUNC/BUSCO to filter contaminated references. |
Metagenomic Sequencing and Analysis Workflow
The following diagram illustrates the end-to-end process of a metagenomic sequencing experiment, highlighting critical steps where issues frequently arise.
The integration of PCR and metagenomic sequencing into the microscopic identification of protozoans creates a powerful, multi-faceted approach to quality control. While PCR offers a targeted, sensitive method for confirming the presence of specific pathogens, mNGS provides a hypothesis-free, comprehensive view of the entire microbial community. By understanding and systematically troubleshooting the common pitfalls outlined in this guide—from optimizing PCR conditions for GC-rich protozoan genomes to selecting and curating appropriate reference databases for metagenomic classification—researchers can significantly enhance the accuracy, reproducibility, and translational impact of their findings.
This technical support center provides troubleshooting guides and FAQs to help researchers navigate the challenges of microscopic identification of protozoans, ensuring robust quality control throughout the experimental workflow.
Frequently Asked Questions & Troubleshooting Guides
Image Acquisition and Handling
Q: My image files are too large and in proprietary formats, making them difficult to share and analyze. What are the best practices for handling this?
A: This is a common challenge in quantitative microscopy.
- Recommended Action: When exporting images from your microscope software, select the TIFF format to avoid "lossy" compression that can introduce artifactual shapes or colors. Ensure export settings are configured to handle the full bit-depth of your data (e.g., 16-bit) to prevent clipping or compression of intensity values. Create a data management plan early in your project to address storage, computational needs, and long-term archiving [4].
Q: How can I ensure my images are suitable for automated analysis later?
A: Image quality at the acquisition stage is critical for downstream analysis.
- Recommended Action: "Keep analysis in mind from the very beginning." Optimize your sample preparation and image acquisition settings during pilot experiments. Incorporate analysis testing from the earliest stages to ensure the images you generate can answer your scientific question [4].
Image Pre-processing and Analysis
Q: The objects of interest in my images have low contrast and are difficult to segment reliably. What can I do?
A: Both classical and deep learning approaches are available.
- Recommended Action: For classical computer vision techniques, pre-processing steps like denoising or deconvolution may be necessary to enhance features so that objects are bright and the background is dark. If this is onerous, consider deep learning segmentation. Deep neural networks can be trained to overcome challenges like debris, variable staining, and low contrast, even in complex biomedical samples [45] [4].
Q: Should I use object detection or instance segmentation for my analysis?
A: The choice depends on your scientific question.
- Recommended Action: Use object detection (which provides a centroid and bounding box) for tasks focused on counting and classification (e.g., "How many cells are infected?"). Use instance segmentation (which finds the exact boundary of each object) when you need to measure object-specific properties (e.g., "What is the size and shape of each infected cell?"). Segmentation is more precise but also a more complex problem [4].
Measurement and Interpretation
Q: What is the appropriate unit for statistical analysis of my image-based data?
A: Determining the unit of comparison is a crucial step.
- Recommended Action: Carefully consider the design of your experiment. The appropriate unit could be an individual object (e.g., a single parasite), a whole image (e.g., a field of view), a biological replicate, or an entire organism. Consistency in this choice is key to valid statistical testing [4].
Experimental Protocols for Key Methodologies
Protocol 1: Real-Time Protozoa Detection Using Deep Learning (YOLOv4)
This protocol outlines the steps for training a deep learning model to automatically detect and classify protozoa in microscopic images [17].
- Dataset Curation: Collect microscopic images from various freshwater sources (e.g., rainwater, puddles). Create a curated dataset containing objects of the target protozoa species (e.g., Bdelloid Rotifera, Paramecium).
- Image Annotation: Manually label each protozoan in the images, drawing bounding boxes around each organism and assigning the correct class label. This annotated dataset is used to train the model.
- Model Training: Implement the YOLOv4 network architecture. Use a framework like PyTorch or TensorFlow. Training parameters from the cited study include:
- Performance Evaluation: Validate the trained model on a held-out test set of images. Calculate standard metrics such as F1-score, precision, sensitivity, and mean Average Precision (mAP) to assess performance [17].
- Deployment: Integrate the trained model into a user-friendly desktop application to allow researchers to test the model on new images [17].
Protocol 2: Establishment of Intestinal Organoid-Derived Monolayers (ODMs) for Infection Studies
This protocol describes the generation of a biologically relevant in vitro platform to study the interaction of protozoan parasites with the intestinal epithelium [46].
- 3D Organoid Culture: Isolate intestinal crypts from the host species of interest (e.g., human, mouse, pig). Embed the crypts in an extracellular matrix (e.g., Matrigel) and culture them in a specialized medium containing growth factors (e.g., EGF, Noggin, R-spondin) to generate and maintain 3D organoids.
- Monolayer Generation: Fragment the 3D organoids into single cells or small clumps. Seed these fragments onto a transwell filter insert pre-coated with a substrate suitable for cell attachment.
- Monolayer Differentiation: Culture the cells on the transwell until they form a confluent, polarized monolayer. The medium can be adjusted to promote differentiation of the various intestinal epithelial cell types.
- Quality Control: Characterize the integrity and functional state of the ODMs before infection. Key methods include:
- Transepithelial Electrical Resistance (TEER): Measure TEER to confirm the formation of tight junctions and an intact barrier.
- Immunolabeling: Use antibodies against marker proteins (e.g., mucin for goblet cells) to verify cell differentiation.
- qRT-PCR: Analyze the transcriptional abundance of tight junction components.
- Infection: Apply the protozoan parasite (e.g., Toxoplasma gondii tachyzoites, Giardia duodenalis trophozoites) to the apical side of the monolayer to initiate the infection study.
Quantitative Data from Cited Experiments
The following table summarizes the performance metrics achieved by the YOLOv4-based protozoa detection framework as described in the search results [17].
Table 1: Performance Metrics for Deep Learning-Based Protozoa Detection
| Metric | Value | Description |
|---|---|---|
| Accuracy | 97% | Overall correctness of the model's predictions. |
| mAP | 0.9752 | Mean Average Precision; overall detection performance. |
| F1-Score | 0.95 | Harmonic mean of precision and sensitivity. |
| Precision | 0.92 | Proportion of correct positive identifications. |
| Sensitivity | 0.98 | Proportion of actual positives correctly identified. |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Featured Experimental Protocols
| Reagent / Material | Function | Example Use Case |
|---|---|---|
| Organoid Culture Medium | Supports growth and differentiation of stem cell-derived intestinal organoids. | Maintaining 3D organoid cultures from human, mouse, pig, and chicken [46]. |
| Extracellular Matrix (e.g., Matrigel) | Provides a scaffold for 3D cell growth, mimicking the basal lamina. | Generation and maintenance of intestinal organoids in vitro [46]. |
| Transwell Filter Inserts | Creates a compartmentalized system for generating polarized epithelial monolayers. | Forming organoid-derived monolayers (ODMs) for apical infection studies [46]. |
| dSTORM Imaging Buffer | Induces fluorophore "blinking" for super-resolution localization. | Enabling dSTORM super-resolution microscopy for nanoscale imaging [45]. |
| Protargol Stain | Silver-based stain that visualizes ciliary and nuclear structures. | Essential for identifying and characterizing ciliates and flagellates based on infraciliary patterns [15]. |
Workflow Visualization with DOT Scripts
Below are diagrams and their corresponding DOT scripts that visualize key workflows and logical relationships in protozoan research.
Sample to Result Workflow
Analysis Method Decision Tree
Protozoa Identification Techniques
Troubleshooting Common Pitfalls and Optimizing Analytical Performance
FAQs: Troubleshooting Common Pre-Analytical Issues
1. Why do my stool samples show degraded protozoan trophozoites, even when preserved? This is often due to a delay between specimen passage and preservation. Trophozoites are particularly labile and begin to degrade quickly. For optimal morphology, fresh stool must be examined, processed, or preserved immediately [47]. If you are using Polyvinyl-Alcohol (PVA), ensure it is a low-viscosity formula designed for optimal preservation of protozoan trophozoites and cysts for permanent staining [47].
2. My parasite egg recovery rates are low from formed stool. What might be the cause? When preserving formed stool in fixatives like 10% formalin or Sodium Acetate-Acetic Acid-Formalin (SAF), it is critical to break the stool up thoroughly and mix it well with the preservative [47]. Inadequate mixing prevents proper fixation throughout the sample, leading to poor recovery during concentration procedures.
3. What is the impact of patient medication on stool specimen analysis? Several drugs and compounds can interfere with analysis, making specimens unsatisfactory for examination. These include:
- Barium or bismuth: Requires a 7-10 day clearance period.
- Antimicrobial agents: Requires a 2-3 week clearance period.
- Mineral oil, antacids, and non-absorbable antidiarrheal preparations. Specimens should be collected before these substances are administered, or collection must be delayed until after the effects have passed [47].
4. How should I handle a stool sample if I need both morphological and molecular data? No single preservative is ideal for all techniques. The recommended practice is to preserve the specimen in two different vials: one with 10% formalin (suitable for concentration procedures and some immunoassays) and another with Low-Viscosity PVA (optimal for permanent stained smears for morphology) [47]. Always confirm the compatibility of your chosen preservative with downstream molecular tests, as some, like formalin, can interfere with PCR, especially after extended fixation [47].
5. Why is the timing of sample collection so critical for some parasitic infections? Many parasites exhibit periodicity, meaning the presence of diagnostic stages (e.g., microfilariae in blood or eggs in urine) fluctuates predictably throughout the day. Collecting samples at the wrong time can lead to false negatives. For example, optimal detection of Wuchereria bancrofti microfilariae is around midnight, while Schistosoma haematobium egg excretion in urine peaks between noon and 3 p.m. [48].
Table 1: Stool Sample Transport and Preservation Guidelines
| Specimen Consistency | Max Transport Time (Unpreserved) | Storage Temp (Unpreserved) | Common Preservatives | Primary Use of Preservative |
|---|---|---|---|---|
| Liquid | ≤ 30 minutes | Room Temperature | SAF, Schaudinn's, PVA | Preservation of trophozoites [48] [47] |
| Semisolid | ≤ 1 hour | Room Temperature | 10% Formalin, SAF, PVA | General purpose; concentration procedures [48] [47] |
| Formed | ≤ 24 hours | 4°C | 10% Formalin, SAF, PVA | General purpose; concentration procedures [48] [47] |
Table 2: Advantages and Disadvantages of Common Stool Preservatives
| Preservative | Advantages | Disadvantages |
|---|---|---|
| 10% Formalin | Good for helminth eggs/larvae; suitable for concentration and immunoassays; long shelf life [47] | Poor for trophozoite morphology; can interfere with PCR; not ideal for permanent stained smears [47] |
| Low-Viscosity PVA (LV-PVA) | Excellent for protozoan trophozoites/cysts; ideal for permanent stained smears (e.g., trichrome) [47] | Contains toxic mercuric chloride; not for concentration; not for acid-fast stains [47] |
| SAF | Suitable for concentration and permanent stains; no mercury; good for acid-fast stains [47] | Requires additive for slide adhesion; permanent stains not as high quality as with PVA [47] |
| Schaudinn's Fixative | Excellent for protozoan trophozoites/cysts; good for permanent stained smears [47] | Contains mercuric chloride; less suitable for concentration procedures [47] |
Experimental Protocols for Sample Processing
Protocol 1: Standard Fixation of Stool Specimens for Comprehensive Analysis
Principle: To ensure specimens are adequate for both morphological identification and potential molecular assays. Procedure:
- Collection: Collect stool in a clean, dry, leak-proof container. Take care to avoid contamination with urine, water, or soil [47].
- Preservation: For a comprehensive approach, preserve the specimen in two vials:
- Vial 1 (10% Formalin): Add 1 volume of stool to 3 volumes of 10% formalin. Mix thoroughly to ensure full fixation. This vial is for concentration procedures and observing eggs, larvae, and cysts [47].
- Vial 2 (LV-PVA): Add 1 volume of stool to 3 volumes of LV-PVA. Mix thoroughly. This vial is for preparing permanent stained smears (e.g., trichrome) for definitive protozoan identification [47].
- Storage: Label vials clearly. Preserved specimens can be stored at room temperature for several months [47].
Protocol 2: Fixation of Cell Monolayers for Transmission Electron Microscopy (TEM)
Principle: To ultrastructurally preserve protozoans cultured in monolayers for high-resolution imaging. Procedure:
- Preparation: Remove the culture medium and wash the monolayer with an appropriate buffer (e.g., 0.1M cacodylate or phosphate buffer) to remove excess protein [49].
- Primary Fixation: Flood the monolayer with a primary fixative, typically 2.5% glutaraldehyde in 0.1M buffer (e.g., phosphate buffer, pH 7.4). Fix at room temperature for 1 hour [49].
- Washing: Aspirate the fixative and wash the cells with buffer 3 times for 5 minutes each.
- Post-Fixation: Osmicate the cells in situ with a 1% osmium tetroxide solution in buffer.
- Scraping: Use a Parafilm-coated spatula to gently scrape the fixed cells from the support surface. Process the resulting suspension as a pellet for further TEM steps (dehydration, embedding, sectioning) [49].
Workflow Visualization: Pre-Analytical Phase
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Sample Fixation and Storage
| Reagent / Material | Function | Application Notes |
|---|---|---|
| 10% Formalin | All-purpose fixative; cross-links proteins. | Preserves helminth eggs, larvae, and protozoan cysts well. Suitable for concentration procedures and some immunoassays [47]. |
| Low-Viscosity PVA (LV-PVA) | Fixative and adhesive; preserves microscopic structure. | Mercury-based; optimal for preparing permanent stained smears for protozoan trophozoite and cyst identification [47]. |
| SAF Solution | Fixative (Sodium Acetate-Acetic Acid-Formalin). | Mercury-free alternative; suitable for concentration, permanent stains, and acid-fast staining [47]. |
| Glutaraldehyde (EM Grade) | Cross-linking fixative for ultrastructural preservation. | Used for Transmission Electron Microscopy (TEM). Provides excellent preservation of cellular detail [49]. |
| 0.1M Phosphate Buffer (pH 7.4) | Isotonic buffer for fixative solutions. | Maintains physiological pH during fixation, preventing artifacts in cellular structure [49]. |
| Pinworm Paddle Kit | Non-invasive collection of perianal specimens. | Used for diagnosing Enterobius vermicularis and Taenia spp. eggs. Best used at night or upon waking [48]. |
Overcoming Technical Hurdles in DNA Extraction from Robust Oocysts and Cysts
Troubleshooting Guide: Common DNA Extraction Issues & Solutions
This guide addresses the most frequent challenges researchers face when extracting DNA from tough-walled protozoan oocysts and cysts, such as those of Cryptosporidium, Giardia, and Entamoeba histolytica.
Table 1: Troubleshooting Common DNA Extraction Problems
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| Low DNA Yield [50] [51] | Inefficient lysis of robust cyst/oocyst walls; DNA loss during purification. | Implement bead-beating or freeze-thaw cycles [50] [51]; Use a smaller elution volume (50-100 µL) [50]; Combine chemical and mechanical lysis methods. |
| PCR Inhibition [50] [51] | Co-purification of inhibitors (e.g., bile salts, complex carbohydrates) from feces. | Use InhibitEX tablets or similar compounds [50]; Add Bovine Serum Albumin (BSA) to PCR reactions [51]; Perform a 1:10 or 1:100 dilution of DNA template prior to PCR [50]. |
| Inconsistent Results Between Samples | Variable cyst/oocyst count; irregular shedding of parasites in feces [20]. | Purify and concentrate cysts from fecal matrix using sucrose flotation or formol-ether [50]; Analyze multiple samples collected on alternate days [20] [52]. |
| Poor DNA Purity (Low A260/A230) [51] | Contamination by organic solvents or carbohydrates from extraction process. | Ensure complete removal of supernatant in wash steps; Use commercial kits like QIAamp DNA Stool Mini Kit, which showed better purity in comparative studies [51]. |
Frequently Asked Questions (FAQs)
FAQ 1: What is the most critical step for successful DNA extraction from robust cysts?
The most critical step is the complete disruption of the robust cyst or oocyst wall. These walls are designed to protect the genetic material in harsh environments, making lysis challenging. A protocol combining mechanical disruption (e.g., bead-beating or freeze-thaw cycles) with chemical and heat lysis (e.g., boiling for 10 minutes) is significantly more effective than relying on a single method [50] [51].
FAQ 2: How can I quickly determine if my DNA extract contains PCR inhibitors?
A reliable method is to spike a known amount of target DNA into your PCR reaction alongside the DNA extract. Failure to amplify the spiked control indicates the presence of inhibitors. Alternatively, you can use broad-range universal primers (e.g., for 16S rDNA) to check for the presence of amplifiable DNA in the sample [50].
FAQ 3: Are commercial DNA extraction kits better than in-house methods for fecal samples?
The choice depends on your priorities. Commercial kits (e.g., QIAamp DNA Stool Mini Kit) are optimized for removing PCR inhibitors and provide consistent results with less hands-on time [50] [51]. However, in-house methods like the Phenol-Chloroform Isoamyl Alcohol (PCI) method can sometimes yield higher DNA concentrations and can be more cost-effective, though they are more labor-intensive and may carry over more inhibitors [51]. One study found the PCI method to have higher diagnostic sensitivity (70%) for Giardia compared to a commercial kit (60%) [51].
FAQ 4: How many stool samples should be processed per patient for reliable molecular detection?
Due to the irregular shedding of parasites, a single stool sample is often insufficient. Traditional microscopy recommends three samples collected on alternate days for optimal sensitivity [20] [52]. However, the high sensitivity of real-time PCR may allow for a reduction in the number of samples needed. One study found that a single stool sample analyzed by a combination of microscopy and real-time PCR was nearly as sensitive as examining three samples by microscopy alone [52].
Experimental Protocols for Key Scenarios
Protocol 1: Optimized DNA Extraction using a Commercial Kit
This protocol is an amendment to the manufacturer's instructions for the QIAamp DNA Stool Mini Kit, specifically designed to improve the recovery of Cryptosporidium DNA [50].
Workflow Overview
Key Reagents & Materials
- QIAamp DNA Stool Mini Kit (Qiagen): Provides lysis buffer, InhibitEX tablets, wash buffers, and silica-membrane columns [50].
- Water Bath or Heat Block: Capable of maintaining 100°C for the extended boiling step [50].
- Pre-cooled Ethanol: Used in the elution step to increase DNA precipitation efficiency [50].
Protocol 2: Phenol-Chloroform Isoamyl Alcohol (PCI) DNA Extraction
A traditional in-house method that can yield high DNA concentration, suitable for situations where cost is a primary concern [51].
Workflow Overview
Key Reagents & Materials
- Sucrose Solution (1M): For the initial purification and concentration of cysts from the fecal debris [51].
- Phenol-Chloroform-Isoamyl Alcohol (25:24:1): A denaturing solution to separate proteins from nucleic acids. Caution: Handle with appropriate PPE as it is toxic. [51]
- Liquid Nitrogen: Used for the freeze cycles in the mechanical disruption step [51].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Materials for DNA Extraction from Oocysts/Cysts
| Reagent/Material | Function in the Protocol |
|---|---|
| InhibitEX Tablets / BSA | Critical for adsorbing and neutralizing PCR inhibitors (bile salts, heme, polysaccharides) present in fecal samples [50] [51] [52]. |
| Silica-Membrane Columns | The core of many commercial kits; allows for selective binding of DNA in the presence of chaotropic salts, followed by washing and elution of pure DNA [50]. |
| Phenol-Chloroform-Isoamyl Alcohol (PCI) | Used in in-house methods to denature and remove proteins from the nucleic acid solution through phase separation [51]. |
| Glass Beads & Bead Beater | Provides mechanical shearing force to physically break open the tough cyst and oocyst walls, complementing chemical lysis [51]. |
| Sucrose Gradient Solution | Used in flotation techniques to purify and concentrate oocysts/cysts away from the bulk of inhibitory fecal material prior to DNA extraction [50] [51]. |
Core Concepts & Frequently Asked Questions (FAQs)
FAQ 1: What is a confidence threshold, and why is adjusting it critical for protozoan detection?
In AI-based classification, a confidence threshold is the minimum probability value a model must assign to a prediction for that result to be accepted. Predictions falling below this threshold are typically rejected and flagged for manual review. Adjusting this threshold is not merely a technical step but a fundamental quality control measure. It allows laboratories to balance accuracy and workflow efficiency, ensuring that the automated system's output meets the specific diagnostic requirements for protozoan identification, where morphological similarities between species can lead to misclassification [53] [54].
FAQ 2: How do I know if my classifier's confidence threshold needs adjustment?
You should investigate threshold adjustment if you observe any of the following in your validation studies:
- High false positive rates: The system is flagging too many artifacts or non-pathogenic protozoa as positive.
- High false negative rates: Manual reviews consistently find organisms the AI missed.
- Inconsistent performance across species: The model performs well for some protozoa (e.g., Giardia cysts) but poorly for others (e.g., Entamoeba species) [53].
These issues indicate that the default threshold may not be optimal for your specific sample preparation methods or the prevalence of certain parasites in your patient population [53].
FAQ 3: What is the trade-off between accuracy and coverage when raising the threshold?
Implementing a confidence threshold creates a direct trade-off between the accuracy of the accepted predictions and the proportion of the data that is automatically labeled (coverage). A higher confidence threshold yields higher accuracy on the accepted predictions but results in a larger number of rejected samples that require manual review.
Recent research demonstrates this clearly: a model with an initial accuracy of 86% could achieve over 95% accuracy by rejecting about 40% of its predictions, or even exceed 99% accuracy by rejecting about 65% of them [54]. This allows each laboratory to choose a threshold that aligns with its diagnostic confidence requirements and available human resources.
Table 1: Impact of Confidence Threshold on Model Performance
| Confidence Threshold | Resulting Accuracy | Data Coverage | Recommended Use Case |
|---|---|---|---|
| Low | Lower (e.g., 86%) | High (e.g., 100%) | Initial screening; high-sensitivity rule-out |
| Medium | High (e.g., >95%) | Moderate (e.g., 60%) | Balanced routine workflow [54] |
| High | Very High (e.g., >99%) | Low (e.g., 35%) | Confirmatory testing; accuracy-critical research [54] |
FAQ 4: Can I use a single confidence threshold for all protozoan species?
No. Using a single, global confidence threshold for all classifiers is not recommended. Different protozoans have distinct morphological characteristics, prevalence, and potential for confusion with artifacts or other species. For instance, a study found that the confidence threshold required adjustment specifically for Schistosoma mansoni to achieve optimal slide-level agreement [53]. The optimal threshold for a large, distinct cyst like Giardia will likely be different from that for a smaller, more variable one like Entamoeba.
Troubleshooting Guide: Common Issues and Solutions
Problem: High False Positive Rate for a Specific Classifier
Scenario: Your AI system is frequently misclassifying artifacts or non-pathogenic protozoa (e.g., Entamoeba coli) as pathogenic species (e.g., Entamoeba histolytica).
Investigation & Solution:
- Confirm Misidentification: Perform a manual review of the false positive cases to verify the nature of the misclassification.
- Analyze Confidence Scores: Export the confidence scores for the incorrect predictions. If they are consistently clustered just above your current threshold, it is a strong indicator that the threshold needs to be raised.
- Adjust Threshold Incrementally: Raise the confidence threshold for the specific problematic classifier (e.g., the E. histolytica classifier) in small steps (e.g., 0.05).
- Re-validate: Use a validated set of images to assess the new False Positive Rate and True Positive Rate at the new threshold. The goal is to find a point where false positives are minimized without significantly impacting the detection of true positives [54].
Problem: High False Negative Rate for a Specific Classifier
Scenario: The system is failing to detect true Blastocystis spp., which are subsequently identified during manual review.
Investigation & Solution:
- Review Image Quality: Ensure that the false negatives are not due to poor image focus, staining, or debris obscuring the organism.
- Inspect Confidence Distribution: Check the confidence scores the model assigned to the missed organisms (the false negatives). If these scores are just below the current threshold, lowering the threshold may be beneficial.
- Lower Threshold Cautiously: Reduce the confidence threshold for that specific classifier incrementally.
- Monitor Impact: Validate that the lowered threshold successfully captures the previously missed targets without introducing an unacceptable number of false positives [53] [54].
Problem: Inconsistent Performance Across Different Sample Preparations
Scenario: Your lab uses multiple fixatives (e.g., SAF, PVA) and you notice the AI performance degrades on samples from one fixative type.
Solution: This is often a "domain shift" issue. The model was likely trained on images from a specific set of preparations. The solution involves:
- Site-Specific Validation: Extensive validation is required for each sample processing method used in your laboratory [53].
- Create Separate Threshold Profiles: You may need to establish and save different confidence threshold settings for each major sample preparation protocol you use. For example, you might have a "Profile A" for SAF-fixed samples and a "Profile B" for MIF-stained samples.
Experimental Protocol: Validating Confidence Thresholds
This protocol provides a step-by-step methodology for establishing and validating organism-specific confidence thresholds in a clinical or research setting.
Objective: To determine the optimal confidence threshold for each protozoan classifier in an AI-based detection system, ensuring maximum accuracy and efficient workflow integration.
Materials & Reagents:
- Validated Image Set: A curated and expertly annotated set of digital microscopic images representing all target protozoan species and common artifacts. This set should be independent of the model's training data.
- AI Classification Software: The platform running the convolutional neural network (CNN) model (e.g., Techcyte Human Fecal Wet Mount algorithm [55] [53]).
- Data Analysis Tool: Software capable of statistical analysis and visualization (e.g., Python with pandas/scikit-learn, R, Excel).
Procedure:
- Image Set Preparation: Assemble your validation set of digital slides. Ensure each image has a confirmed "ground truth" label assigned by an expert microscopist.
- Baseline Model Inference: Run the entire validation set through your AI model without any post-filtering. Export the results, including the predicted class and the model's confidence score (a value between 0 and 1) for each object detected.
- Performance Analysis: For each protozoan classifier (e.g., the Giardia cyst classifier), perform the following:
- Sort all predictions for that class by confidence score.
- Calculate accuracy metrics (True Positives, False Positives, False Negatives) at various confidence thresholds (e.g., from 0.5 to 0.95 in 0.05 increments).
- For each threshold, compute the accuracy (of accepted predictions) and coverage (percentage of total predictions that are accepted) [54].
- Generate Accuracy/Coverage Curves: Plot the results for each classifier on a graph, with coverage on the X-axis and accuracy on the Y-axis. This visualization makes the trade-off clear and facilitates threshold selection.
- Threshold Selection: Convene a review with senior staff. Based on the accuracy/coverage curves and your laboratory's operational capacity for manual review, select an optimal confidence threshold for each classifier. For example, you might decide that for the critical pathogen Entamoeba histolytica, you require 99% accuracy, while for Blastocystis spp., 95% accuracy is acceptable [54].
- Implementation and Documentation: Configure your AI system with the new, fine-tuned thresholds. Document the entire process, including the rationale for each chosen threshold, in your quality control records.
The Scientist's Toolkit: Research Reagent Solutions
The following reagents and materials are essential for preparing samples for AI-based microscopic analysis, as cited in recent validation studies.
Table 2: Essential Materials for Wet-Mount Parasitology Analysis
| Reagent / Material | Function / Description | Example Use in Protocol |
|---|---|---|
| SAF Fixative(Sodium-Acetate-Acetic Acid-Formalin) | Preserves morphological integrity of parasites during transport and processing [53]. | Used as the primary fixative in tubes for stool samples [53]. |
| StorAX SAF Filtration Device | A proprietary system for concentrating parasitic structures from SAF-fixed samples [53]. | Used for sample concentration via filtration and centrifugation [53]. |
| Lugol's Iodine Solution | A common staining solution that enhances contrast of protozoan cysts and nuclei [53]. | Mixed with glycerol/PBS as a mounting medium for wet-mount slides [53]. |
| Mounting Medium(Lugol's Iodine & Glycerol in PBS) | Final medium for slide preparation; iodine stains structures, glycerol preserves and prevents drying [53]. | 15µL of sediment mixed with 15µL of mounting medium for slide preparation [53]. |
| Ethyl Acetate | Used in concentration techniques to extract fat and debris from the sample, cleaning the final sediment [53]. | Added during the concentration step prior to centrifugation [53]. |
| Triton X-100 | A detergent used to enhance the release of parasitic elements from the stool matrix [53]. | Added during the sample homogenization and filtration process [53]. |
Managing Image Quality and Handling Difficult Specimens for Consistent AI Performance
Troubleshooting Guides
Guide 1: Addressing Poor Contrast in Unstained Protozoan Specimens
Problem: AI models fail to reliably detect or classify unstained, transparent protozoans in brightfield microscopy due to insufficient contrast.
Explanation: Unstained biological specimens are primarily "phase objects;" they cause a phase shift in light passing through them but minimal change in amplitude (brightness), making them nearly invisible to human eyes and cameras in standard brightfield mode [56]. Since AI models are trained on image features, this lack of discernible features leads to poor performance.
Solution: Employ optical contrast techniques tailored to your specimen type.
- For thin, unstained specimens (e.g., live culture cells): Use Phase Contrast microscopy. This technique translates minute phase shifts into observable amplitude shifts, making transparent structures visible [57].
- For observing internal details and 3D structure: Use Differential Interference Contrast (DIC). DIC provides an apparent 3D shadowed image and does not produce the bright "halo" artifacts common in phase contrast [57] [58].
Protocol: Setting Up Phase Contrast Microscopy
- Requirement: A microscope equipped with a phase condenser and phase contrast objectives (inscribed with Ph1, Ph2, or Ph3).
- Steps:
- Rotate the condenser turret to the position that matches your objective (e.g., Ph2 for a Ph2 objective).
- Install a centering telescope in place of an eyepiece.
- While looking through the centering telescope, bring the bright phase ring (in the condenser) and the dark phase plate (inside the objective) into focus simultaneously.
- Use the adjustment wrenches on the condenser to center the bright ring so it perfectly overlaps the dark ring [57].
- Remove the centering telescope and resume observation.
Guide 2: Managing AI Errors Caused by Artifacts and Specimen Variability
Problem: AI model performance degrades due to imaging artifacts, variable staining, debris, or overlapping structures in thick samples.
Explanation: AI models, especially those trained on limited or idealized datasets, can be confused by features not present in their training data. Halo artifacts from phase contrast, out-of-focus information in thick specimens, and floating debris can be misinterpreted as protozoan features [59] [57]. This is a form of "domain shift" where the real-world data differs from the training data [60].
Solution: Optimize sample preparation and leverage advanced AI training strategies.
- Minimize Thickness: For thick specimens, create thin smears or sections to reduce overlapping structures and out-of-focus light that cause halos and blurred features [57].
- Clean Protocols: Ensure careful sample washing and preparation to minimize debris.
- Data Augmentation: Train AI models using augmented datasets that include simulated artifacts, variations in staining intensity, and synthetic data to improve robustness [61].
- Realistic Simulation: Use simulation platforms like pySTED to generate large volumes of realistic training data with controlled artifacts, which is particularly useful for super-resolution techniques [61].
Guide 3: Ensuring Microscope Performance for Quantitative AI Analysis
Problem: Inconsistent AI performance over time due to drift in microscope optical performance, such as declining light source intensity or lens misalignment.
Explanation: Microscopes are complex systems whose performance can waver, introducing bias into images. AI models that rely on precise measurements of intensity or morphology will produce unreliable results if the imaging system itself is unstable [62]. This affects the reproducibility of experiments.
Solution: Implement a regular microscope quality control (QC) program.
- Use QC Tools: Employ commercial quality control slides (e.g., from Argolight) that contain precise fluorescent patterns. These slides are imaged periodically, and the images are analyzed by companion software to check key parameters like illumination homogeneity, resolution, and stage positioning accuracy [62].
- Built-in Monitoring: Utilize modern microscopes with built-in performance monitoring. For example, some confocal systems have laser intensity monitoring and feedback control to ensure stable excitation during long time-lapse experiments [62].
Protocol: Basic Microscope Performance Check
- Requirement: An Argolight slide or similar QC tool.
- Steps:
- Image the QC slide (e.g., the "field of rings" pattern for flatness, "gradually spaced lines" for resolution) using your standard imaging protocol.
- Analyze the image with the designated software.
- Review the automated report detailing performance metrics for each parameter.
- If any metric falls outside your acceptable range, generate a service report for your facility manager or manufacturer to streamline troubleshooting [62].
Frequently Asked Questions (FAQs)
FAQ 1: What are the best deep learning architectures for protozoan detection and classification? The optimal architecture depends on your specific task:
- For real-time detection of multiple protozoans in a video or image: The YOLO (You Only Look Once) series is state-of-the-art, known for its speed and accuracy. Modifications of YOLOv3/YOLOv4 have been successfully used to detect Plasmodium in images captured via smartphone [59] [17].
- For pixel-level segmentation to observe parasite morphology: U-Net-based models are widely used and highly effective [59] [61].
- For image classification (e.g., identifying parasite species): Convolutional Neural Networks (CNNs) are typically employed. Advanced networks like Attentive Dense Circular Net (ADCN) have achieved very high accuracy (>97%) in classifying infected red blood cells [59].
FAQ 2: How can I overcome the challenge of limited annotated data for training AI models? Several strategies can mitigate the need for vast, hand-labeled datasets:
- Weakly Supervised Learning: Use sample-level labels (e.g., "image contains Plasmodium") instead of costly bounding boxes or pixel-level annotations. Methods like Multiple Objects Features Fusion (MOFF) have shown success with this approach [59].
- Unsupervised Learning: Models like Graph Convolutional Networks (GCNs) can recognize parasite stages without any image labels, achieving high accuracy by learning the data's inherent structure [59].
- Transfer Learning: Fine-tune a pre-trained model (trained on a large general dataset) on your smaller, specific protozoan dataset [59].
- Synthetic Data Generation: Use realistic simulation platforms to generate unlimited, perfectly annotated training data, as demonstrated with pySTED for super-resolution microscopy [61].
FAQ 3: My AI model works well in brightfield but fails in phase contrast images. Why? This is a classic domain shift problem. Features learned by the model from brightfield images (e.g., based on color and absorption) are not directly transferable to phase contrast images, which are dominated by edge effects and halos [57] [60]. To fix this:
- Re-train the Model: Fine-tune your model using a dataset that includes phase contrast images.
- Data Augmentation: Augment your brightfield training set with synthetic phase contrast artifacts to make the model more robust.
- Multi-Modal Training: Train a single model on a combined dataset containing both brightfield and phase contrast images from the start.
Data Presentation
Table 1: Comparison of Optical Contrast Methods for Protozoan Imaging
| Technique | Best For | Key Advantages | Key Limitations | Impact on AI Performance |
|---|---|---|---|---|
| Brightfield [57] [56] | Stained, colored, or thick specimens. | Simple setup, true color representation. | Very low contrast for unstained, transparent specimens. | Poor performance on live/unstained samples due to lack of features. |
| Phase Contrast [57] [56] | Thin, unstained specimens (e.g., live cultures). | Excellent for observing internal structures of live cells. | Produces bright "halo" artifacts that can obscure details, especially in thick samples. | Halos can be misinterpreted by AI unless training data includes such artifacts. |
| DIC [57] [58] | Unstained specimens, highlighting 3D structure. | High-resolution, optical "sectioning" reduces out-of-focus blur. No halo artifacts. | More complex and expensive setup. Not suitable for plastic dishes. | Provides high-contrast, detailed images that can improve segmentation and morphology models. |
| Darkfield [57] | Detecting very small organisms or bacteria. | Brilliant contrast on a dark background. | Only reveals object outlines, not internal details. | Useful for detection tasks but not for internal classification. |
Table 2: Key Reagent and Tool Solutions for Quality Control
| Item | Function/Description | Relevance to Consistent AI Performance |
|---|---|---|
| Argolight Slide [62] | A fluorescent slide with precise geometrical patterns (e.g., lines, rings). | Used to quantitatively monitor microscope parameters (homogeneity, resolution) over time, ensuring the input data for AI is stable. |
| Validated Fluorophore (e.g., ATTO-647N) [61] | A fluorescent dye with well-characterized photophysical properties. | Serves as a standard for validating imaging protocols and simulation parameters, crucial for generating reliable ground-truth data. |
| pySTED Simulation Platform [61] | A realistic, open-source Python environment that simulates STED microscopy acquisition. | Generates large, perfectly annotated synthetic datasets to train and benchmark AI models, overcoming the limitation of scarce biological data. |
| U-Netdata map Model [61] | A deep learning model that predicts the underlying structure of a real microscopy image. | Creates realistic data maps from real images, which can be used in simulators to generate synthetic images with different imaging parameters for robust AI training. |
Experimental Protocols & Workflows
Workflow 1: Specimen Preparation and Imaging for AI Analysis
The following diagram outlines a standardized workflow for preparing and imaging protozoan specimens to ensure high-quality, consistent data for AI model training and deployment.
Workflow 2: AI-Assisted Microscopy Feedback Loop
This diagram illustrates a closed-loop framework for using AI not just for analysis, but also to intelligently guide the microscopy acquisition process itself, optimizing for both image quality and data relevance.
Strategies for Differentiating Morphologically Similar Species and Reducing False Positives
Frequently Asked Questions (FAQs)
FAQ 1: Why is microscopic examination still relevant in modern parasitology diagnostics? Despite advances in molecular methods, light microscopy remains the gold standard for many parasitic infections. It is a cost-effective, versatile technique that can detect a wide range of parasites in a single test without requiring prior knowledge of the potential pathogen. It is particularly crucial for identifying helminths and protozoa not covered by commercial multiplex PCR panels and is easily adaptable for resource-poor settings [63].
FAQ 2: What are the main limitations of molecular methods like PCR for parasite identification? Molecular methods have several limitations:
- Insufficient Coverage: Commercial tests are not available for all medically important parasites. Humans can harbor over 90 common parasitic species, but NAATs typically target only a few, such as Giardia, Cryptosporidium, and Entamoeba histolytica [63].
- Specimen Incompatibility: Fecal samples contain PCR inhibitors like bile salts and complex polysaccharides. Furthermore, formalin fixation, commonly used for specimen preservation, rapidly degrades DNA, making molecular analysis difficult or impossible [63].
- Inadequate Databases: Accurate identification via sequencing requires comprehensive reference databases, which are currently incomplete for all human-infecting parasite species, potentially leading to missed or incorrect diagnoses [63].
FAQ 3: How can my lab improve the detection of parasites with robust cyst walls, like Cryptosporidium? A key strategy is implementing a more efficient DNA extraction protocol. Traditional methods like freeze-thaw cycles are time-consuming. A recent metagenomic-next-generation sequencing (mNGS) assay uses a device for rapid microbial lysis (e.g., within 3 minutes), followed by DNA extraction and whole-genome amplification. This method has proven sensitive for detecting as few as 100 Cryptosporidium oocysts on 25g of lettuce and can simultaneously identify multiple protozoan parasites [9].
FAQ 4: What is a common mistake that reduces contrast and resolution during microscopic examination? A common practice to increase image contrast is to reduce the condenser aperture diaphragm or lower the substage condenser. While this maneuver indeed increases contrast, it simultaneously seriously reduces resolution and image sharpness. Control of contrast should be achieved through proper optical techniques and settings rather than compromising the condenser aperture [64].
Troubleshooting Guides
Problem 1: Low Sensitivity in Detecting Protozoan Parasites in Stool Samples
Issue: Traditional microscopy is missing low-intensity infections of pathogenic protozoa.
Solution: Implement a complementary multiplex PCR assay alongside microscopy.
- 1. Identify the Problem: Review diagnostic results and note if clinically suspected cases are consistently microscopy-negative.
- 2. Establish a Theory of Probable Cause: The sensitivity of microscopy may be insufficient for low parasite loads [35].
- 3. Test the Theory & Resolution Plan:
- Plan of Action: Integrate a commercial multiplex real-time PCR (qPCR) assay, such as the AllPlex Gastrointestinal Panel, into the diagnostic workflow. This assay targets protozoa including Giardia intestinalis, Cryptosporidium spp., Entamoeba histolytica, Dientamoeba fragilis, and Blastocystis spp. [35].
- Implementation:
- Suspend fresh stool samples in an appropriate transport medium.
- Extract DNA using an automated extraction system.
- Perform multiplex PCR amplification according to the manufacturer's protocol.
- Analyze amplification curves; any cycle quantification (Cq) value ≤ 40 is typically considered positive.
- 4. Verify System Functionality: A 2025 prospective study demonstrated that multiplex PCR detected significantly more positive samples for key protozoa compared to microscopy, as shown in Table 1 [35].
- 5. Document Findings and Lessons Learned: Maintain records of parallel testing to validate the increased sensitivity of the molecular assay in your specific lab setting. Note that microscopy remains essential for detecting parasites not targeted by the panel, such as Cystoisospora belli and helminths [35].
Problem 2: Differentiating Morphologically Similar Species in Microscopy
Issue: Inability to reliably distinguish between pathogenic and non-pathogenic species that look identical, such as Entamoeba histolytica (pathogenic) and Entamoeba dispar (non-pathogenic).
Solution: Employ a dual-method approach for definitive identification.
- 1. Identify the Problem: Microscopy reveals Entamoeba histolytica/dispar complex cysts or trophozoites, but clinical action requires knowing if the pathogen E. histolytica is present.
- 2. Establish a Theory of Probable Cause: Light microscopy lacks the resolution to differentiate these species based on morphology alone [63].
- 3. Test the Theory & Resolution Plan:
- Plan of Action: Use microscopy for initial genus-level detection and follow up with a species-specific test.
- Implementation:
- Microscopy: Perform microscopic examination with concentration methods on stool samples. Report finding as "Entamoeba histolytica/dispar complex" to indicate the diagnostic uncertainty.
- Molecular Confirmation: Use a PCR test that is specific for Entamoeba histolytica. Multiplex PCR panels can directly distinguish it from other entrants [35]. Alternatively, specific laboratory-developed tests (LDTs) can be used.
- 4. Verify System Functionality: This strategy ensures that pathogenic E. histolytica infections are accurately identified and treated, while non-pathogenic E. dispar infections are correctly reported, preventing unnecessary treatment.
- 5. Document Findings: Update laboratory protocols to reflexively perform PCR on any sample where the E. histolytica/dispar complex is identified by microscopy.
Data Presentation
Table 1: Comparison of Detection Rates for Intestinal Protozoa by Microscopy vs. Multiplex PCR (3,495 Stool Samples) [35]
| Parasite | Microscopy Detection Rate (No. of Positive Samples) | Multiplex PCR Detection Rate (No. of Positive Samples) |
|---|---|---|
| Giardia intestinalis | 0.7% (25) | 1.28% (45) |
| Cryptosporidium spp. | 0.23% (8) | 0.85% (30) |
| Entamoeba histolytica | 0.68% (24) | 0.25% (9) |
| Dientamoeba fragilis | 0.63% (22) | 8.86% (310) |
| Blastocystis spp. | 6.55% (229) | 19.25% (673) |
Note: The higher detection rate for *E. histolytica by microscopy is attributed to the cross-reactivity of microscopy with the non-pathogenic E. dispar. PCR provides specific identification [35].*
Table 2: Strengths and Limitations of Parasite Diagnostic Methods [63]
| Diagnostic Characteristic | Morphology-Based Diagnostics | PCR-Based Diagnostics | Sequencing-Based Diagnostics |
|---|---|---|---|
| Sensitivity | ++ | +++ | +++ |
| Specificity | +++ | +++ | +++ |
| Genus-level ID | +++ | +++ | +++ |
| Species-level ID | ++ | +++ | +++ |
| All parasites in one test | +++ | - | - |
| Detects novel/zoonotic agents | +++ | - | +++ |
| Cost-effectiveness | +++ | ++ | + |
Key: -, no/low capacity/efficacy; +, limited; ++, moderate; +++, high capacity/efficacy.
Experimental Protocols
Protocol: Metagenomic Detection of Protozoan Parasites from Leafy Greens [9]
This protocol describes a method for identifying parasites on fresh produce using metagenomic next-generation sequencing (mNGS).
Sample Preparation and Spiking:
- Place a 25g lettuce leaf in a sterile container.
- Spike the leaf surface with a known quantity of parasite oocysts/cysts (e.g., Cryptosporidium parvum, Giardia duodenalis) in phosphate-buffered saline (PBS). Air-dry for 15 minutes.
Washing and Concentration:
- Transfer the spiked leaf to a stomacher bag with 40ml of buffered peptone water with 0.1% Tween.
- Homogenize in a stomacher at 115 rpm for 1 minute.
- Pass the fluid through a 35μm filter under vacuum to remove plant debris.
- Centrifuge the filtrate at 15,000 x g for 60 minutes at 4°C. Discard the supernatant.
Rapid DNA Extraction and Amplification:
- Lyse the microbial pellet rapidly (e.g., using an OmniLyse device for ~3 minutes).
- Extract DNA via acetate precipitation.
- Subject the extracted DNA to whole-genome amplification to generate sufficient quantities (typically 0.16–8.25 μg) for sequencing.
Sequencing and Bioinformatics:
- Prepare libraries from the amplified DNA.
- Sequence using a nanopore (MinION) or an alternate platform (Ion S5).
- Analyze the generated fastq files using a bioinformatics platform (e.g., CosmosID webserver) for microbial identification.
Workflow Visualization
Differentiation of Entamoeba Species Workflow
mNGS Parasite Detection from Produce
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Advanced Parasite Detection
| Item | Function/Benefit |
|---|---|
| OmniLyse Device | Enables rapid (3-minute) mechanical lysis of robust parasite oocyst/cyst walls, facilitating efficient DNA release for sequencing [9]. |
| Whole Genome Amplification Kits | Amplifies small quantities of extracted DNA to the microgram amounts required for next-generation sequencing library preparation [9]. |
| Multiplex PCR Panels (e.g., AllPlex GIP) | Allows simultaneous detection and differentiation of multiple gastrointestinal protozoa in a single stool sample, increasing throughput and sensitivity [35]. |
| FecalSwab Medium | Preserves nucleic acids in stool samples during transport and storage, making them suitable for subsequent molecular testing [35]. |
| DNA Extraction Kits (for feces) | Designed to efficiently isolate PCR-quality DNA from complex fecal samples while mitigating the effects of common PCR inhibitors [63] [35]. |
| Non-Formalin Fixatives (e.g., 70% Ethanol) | Preserves parasite morphology for potential microscopy while also maintaining DNA integrity for downstream molecular assays, unlike formalin [63]. |
Validation Frameworks and Comparative Analysis of Diagnostic Platforms
Frequently Asked Questions (FAQs)
Q1: What are the key performance parameters I need to validate for a new diagnostic method in protozoan research? The core parameters for validating a new diagnostic method are Accuracy, Precision, and the Limit of Detection (LoD). Accuracy determines how close your results are to the true value, often assessed by comparison to a gold standard method. Precision evaluates the reproducibility of your results under defined conditions. The LoD is the lowest quantity of the parasite that can be reliably detected by your assay [53] [65].
Q2: How do I determine the Limit of Detection (LoD) for a microscopy-based method? The LoD is determined by testing a series of samples with known, decreasing concentrations of the target protozoan. The lowest concentration at which the parasite is detected in ≥95% of replicates is established as the LoD. This often requires creating spiked samples using reference materials, such as a known number of oocysts or cysts [65].
Q3: My molecular assay for Cryptosporidium is showing inconsistent results near the detection limit. What could be wrong? Inconsistent results near the LoD are not uncommon and can be influenced by factors such as inhibitor presence in the stool matrix, extraction efficiency, or primer-binding affinity. It is recommended to perform repeat testing (as described in the troubleshooting guide below) and ensure the target concentration in your quality control samples is well above the established LoD for reliable routine use [65].
Q4: What is considered a good level of agreement between a new method and the gold standard? A strong level of agreement is often indicated by a Cohen's Kappa coefficient (κ) of >0.90, which represents almost perfect agreement. Overall percentage agreement values above 95% are also typically considered excellent, though this can vary by application. One study on an AI-based parasite detection system reported an overall agreement of 98.1% with light microscopy and a κ of 0.915 [53].
Q5: How can I assess the precision of my validation method? Precision is assessed through repeatability (intra-run) and reproducibility (inter-run) studies. This involves testing the same sample multiple times in a single run and across different runs, days, or operators. High precision is demonstrated by minimal variance and consistent results, with high positive and negative percentage agreements [53].
Troubleshooting Guide
Issue 1: Low Analytical Sensitivity (High LoD)
| Observed Problem | Potential Causes | Recommended Actions |
|---|---|---|
| The method fails to consistently detect low concentrations of the target protozoan. | - Suboptimal sample preparation or concentration technique.- Insufficient staining or imaging quality.- PCR inhibitors in the sample matrix (for molecular methods). | - Validate and optimize the sample concentration method (e.g., formalin-ethyl acetate centrifugation) [53].- Use standardized mounting media and verify staining protocols.- For PCR, incorporate an internal control to detect inhibition and dilute samples to mitigate its effects [65]. |
Issue 2: Poor Precision and Reproducibility
| Observed Problem | Potential Causes | Recommended Actions |
|---|---|---|
| Inconsistent results when testing the same sample multiple times. | - Unstandardized manual steps in sample processing.- Variable slide scanning or imaging conditions.- Operator-dependent interpretation. | - Implement Standard Operating Procedures (SOPs) for every manual step.- Use a calibrated slide scanner and standardize focal planes and magnification [53].- For AI-assistance, ensure the algorithm's confidence thresholds are optimized for your lab's specific conditions [53]. |
Issue 3: Discrepant Results Compared to Gold Standard
| Observed Problem | Potential Causes | Recommended Actions |
|---|---|---|
| New method results do not match those from the reference method (e.g., light microscopy). | - The new method may have higher sensitivity for certain organisms (e.g., Blastocystis spp.) [53].- The gold standard may have inherent, operator-dependent variability. | - Perform a discrepant analysis on the samples. Use an additional, validated method (e.g., PCR) to resolve the true positive status [53].- Ensure all technologists using the gold standard are highly trained and, if possible, blinded to the sample status. |
Protocol 1: Determining Limit of Detection (LoD)
This protocol is adapted from the procedure used to validate the BD MAX Enteric Parasite Panel [65].
- Obtain Standard Materials: Acquire quantified reference materials, such as purified oocysts (Cryptosporidium), cysts (Giardia), or genomic DNA (Entamoeba histolytica).
- Prepare Serial Dilutions: Spike negative stool matrix with the standard material to create a series of concentrations, typically in a logarithmic dilution series (e.g., 50,000, 25,000, 12,500 oocysts/mL, etc.).
- Testing Replicates: Test each concentration in duplicate or triplicate in independent assays.
- Establish LoD: The LoD is the lowest concentration at which ≥95% of the test replicates return a positive result. For example, if a concentration of 6,250 oocysts/mL is detected in all replicates, but the next lower dilution is not, this concentration is confirmed as the LoD [65].
Protocol 2: Validating Accuracy and Precision Against a Gold Standard
This protocol follows the design used in the validation of digital microscopy/AI systems [53].
- Sample Collection: Assay a panel of well-characterized samples, including:
- Reference Samples: A panel of confirmed positive (covering all target parasites) and negative samples.
- Prospective Clinical Samples: All routine samples submitted for testing over a defined period (e.g., 3 months).
- Parallel Testing: Process all samples in parallel using both the new method (e.g., Digital Microscopy/CNN) and the established gold standard (e.g., Light Microscopy). Technologists should be blinded to the results of the other method.
- Data Analysis:
- Calculate Positive Percentage Agreement and Negative Percentage Agreement.
- Determine Overall Agreement and Cohen's Kappa statistic to measure agreement beyond chance.
- For precision, perform intra-run (same run) and inter-run (different runs/days) testing on a subset of samples and calculate the percent agreement.
Table 1: Performance Metrics of a Digital Microscopy/AI Workflow for Intestinal Parasite Detection [53]
| Parameter | Result on Reference Samples | Result on Prospective Clinical Samples |
|---|---|---|
| Positive Agreement | 97.6% | - |
| Negative Agreement | 96.0% | - |
| Overall Agreement | - | 98.1% |
| Cohen's Kappa (κ) | - | 0.915 |
Table 2: Limit of Detection (LoD) for a Molecular Assay (BD MAX EPP) [65]
| Target Parasite | LoD |
|---|---|
| Giardia lamblia | 781 cysts/mL |
| Cryptosporidium parvum | 6,250 oocysts/mL |
| Entamoeba histolytica | 125 DNA copies/mL |
Table 3: Performance of Deep Learning Models in Stool Parasite Identification [66]
| Model | Accuracy | Precision | Sensitivity | Specificity | F1-Score |
|---|---|---|---|---|---|
| DINOv2-large | 98.93% | 84.52% | 78.00% | 99.57% | 81.13% |
| YOLOv8-m | 97.59% | 62.02% | 46.78% | 99.13% | 53.33% |
Experimental Workflow Visualization
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Materials for Validation Studies in Protozoan Identification
| Reagent / Material | Function in Validation | Example from Literature |
|---|---|---|
| Quantified Oocysts/Cysts | Serve as standardized reference material for spiking experiments to determine LoD and accuracy. | Giardia lamblia cysts and Cryptosporidium parvum oocysts from Waterborne Inc. were used to prepare simulated stool samples [65]. |
| SAF Fixative | Preserves the morphological integrity of protozoans in stool samples during transport and processing. | Sodium-acetate-acetic acid-formalin (SAF) was used for all stool samples in a digital microscopy validation study [53]. |
| Formalin-Ethyl Acetate | Used in concentration techniques (FECT) to enrich parasitic elements in stool sediments for microscopy. | The Formalin-Ethyl Acetate Centrifugation Technique (FECT) was used as a gold standard method [66]. |
| Mounting Medium (Lugol's Iodine/Glycerol) | Enhances contrast for microscopic visualization of protozoan cysts and trophozoites. | A mounting medium of Lugol's iodine and glycerol in PBS was used for wet-mount slide preparation [53]. |
| Convolutional Neural Network (CNN) Algorithm | AI model for automated detection and pre-classification of parasitic structures in digital images. | The Techcyte Human Fecal Wet Mount (HFW) algorithm was validated for detecting intestinal parasites [53]. |
Troubleshooting Guides
Guide 1: Addressing Low Slide-Level Agreement in AI Model
Problem: The AI model's diagnosis shows poor agreement with manual light microscopy assessments.
| Possible Cause | Diagnostic Steps | Recommended Solution |
|---|---|---|
| Poor Image Quality [67] | Inspect Whole Slide Image (WSI) for blur, artifacts, or improper staining. | Re-scan slides using a calibrated scanner (e.g., NanoZoomer HT2). Standardize PASM staining protocols. [67] |
| Insufficient Model Training [67] [18] | Review model performance metrics (F1-score, precision, recall) on validation cohorts. | Re-train the model using a larger, diverse dataset. Employ data augmentation techniques. Ensure class balance in the training cohort. [67] |
| Domain Shift [67] | Check if images are from a new scanner or staining protocol not seen in training. | Apply domain adaptation techniques. Re-calibrate the model using a small set of images from the new source. |
| Incorrect Glomerular Segmentation [67] | Validate the output of the glomerular localization module (GloSNet). | Manually review segmented glomeruli. Re-train the segmentation network with more annotated data. |
Guide 2: Improving Analytical Sensitivity for Protozoan Detection
Problem: The AI model fails to detect low-density parasitic infections, resulting in false negatives.
| Possible Cause | Diagnostic Steps | Recommended Solution |
|---|---|---|
| Class Imbalance [67] | Analyze the distribution of parasite densities in the training data. | Use oversampling or weighted loss functions during training to focus on low-density examples. |
| Inadequate Resolution [18] | Verify if the image magnification (e.g., 20x, 40x) is sufficient to resolve target protozoa. | Use a higher objective lens (e.g., 40x) for scanning. Ensure the model is trained on high-magnification patches. |
| Subtle Morphological Features [25] | Consult a parasitologist to confirm that diagnostic features are visually distinct. | Incorporate staining techniques (e.g., Giemsa, Protargol) that highlight specific structures. [25] Focus model attention on relevant regions. |
Frequently Asked Questions (FAQs)
Q1: What are the key performance metrics I should use to benchmark my AI model against light microscopy?
You should use a combination of metrics to comprehensively evaluate performance. The table below summarizes the key metrics and their target values based on recent research.
| Metric | Formula/Description | Target Benchmark (from recent studies) |
|---|---|---|
| Slide-Level Agreement | Percentage of whole-slide images where AI and pathologist diagnoses match. | High agreement is crucial; specific benchmarks are study-dependent. [67] |
| F1-Score | Harmonic mean of precision and recall: 2(PrecisionRecall)/(Precision+Recall) | 83.86% - 85.45% (in external validation cohorts) [67] |
| Precision | Proportion of positive identifications that are correct: True Positives/(True Positives+False Positives) | 81.37% - 83.12% (in external validation cohorts) [67] |
| Recall (Sensitivity) | Proportion of actual positives correctly identified: True Positives/(True Positives+False Negatives) | 87.84% - 88.94% (in external validation cohorts) [67] |
| Accuracy | Overall proportion of correct predictions: (True Positives+True Negatives)/Total Predictions | Reported in studies, but should be considered alongside F1 for imbalanced data. [67] |
Q2: My model performs well on internal data but poorly on external data from a different clinic. What is the most likely cause and how can I fix it?
This is a classic domain shift problem. Causes include differences in slide scanners, staining protocols (e.g., variations in PASM staining), or sample preparation methods across institutions. [67]
Solutions:
- Multi-Center Training: Develop and train your model using datasets sourced from multiple institutions with different protocols. [67]
- External Validation: Always include independent external validation cohorts from different centers in your benchmarking study, as demonstrated in the glomerular nephritis study with two external cohorts. [67]
- Stain Normalization: Apply computational techniques to normalize color and intensity variations between images from different sources before analysis.
Q3: What are the best practices for preparing a high-quality image dataset for training an AI model in parasitic protozoan diagnosis?
Best practices involve rigorous standardization at every stage.
- Standardized Staining: Use consistent staining protocols (e.g., Giemsa, PASM) across all samples. [67] [25]
- High-Quality Scanning: Use calibrated whole-slide scanners (e.g., Hamamatsu NanoZoomer) with a fixed magnification (e.g., 20x or 40x objective). [67]
- Expert Annotation: Have all training images annotated and verified by multiple experienced parasitologists to establish a reliable ground truth and minimize inter-observer variability. [67] [2]
- Data Diversity: Ensure the dataset includes images from different parasite life cycles, host species, and density levels, and reflects various imaging conditions. [18] [25]
Q4: How can I visualize and understand what features my AI model is using to make a diagnosis?
This addresses the "black box" problem common in deep learning.
- Attention Maps: Implement visualization techniques like attention hotspots to highlight which regions of the whole-slide image (e.g., specific glomeruli or parasite structures) were most influential in the model's decision. [67]
- Model Interpretation: This remains an active area of research. Future studies are expected to focus more on improving model interpretability for clinical transparency. [67]
Experimental Protocols for Benchmarking
Protocol 1: Slide-Level Agreement Study
Objective: To quantify the diagnostic concordance between the AI model and manual light microscopy by nephropathologists.
Methodology:
- Cohort Selection: Select a minimum of 300 whole-slide images (WSIs) from a biobank, representing a range of protozoan infections and negative cases. [67]
- Blinding: De-identify and randomize all WSIs.
- Manual Assessment: A panel of at least two expert parasitologists will independently examine each WSI via light microscopy and provide a diagnosis. A consensus diagnosis will be established for any discrepancies, serving as the ground truth. [67] [2]
- AI Assessment: Process the same set of WSIs through the AI-assisted diagnostic model to generate an automated diagnosis. [67]
- Statistical Analysis: Calculate the percentage agreement and Cohen's Kappa between the AI diagnosis and the consensus manual diagnosis.
Protocol 2: Analytical Sensitivity (Limit of Detection)
Objective: To determine the lowest density of parasites per unit area that the AI model can reliably detect.
Methodology:
- Sample Preparation: Prepare a dilution series of parasite-positive samples, confirmed by expert microscopy, to create slides with known, decreasing parasite densities. [25]
- Image Acquisition: Scan all slides using a standardized protocol. [67]
- AI Testing: Run the AI model on each WSI in the dilution series. For each slide, record a binary outcome (positive/negative) and the confidence score.
- Data Analysis: Use probit regression analysis to determine the parasite density at which the model achieves 95% detection probability, establishing the limit of detection.
Workflow and Relationship Diagrams
The Scientist's Toolkit: Research Reagent Solutions
| Essential Material | Function in Experiment | Example Use Case |
|---|---|---|
| Periodic Acid-Silver Methenamine (PASM) [67] | Provides optimal contrast for visualizing basement membranes and complex structures in tissue. | Staining kidney biopsy sections for glomerular analysis; can be adapted for certain protozoan structures. [67] |
| Giemsa Stain [25] | Stains nuclear material and cytoplasmic components, helping differentiate cell types and parasite morphology. | Identifying and classifying blood-borne protozoa like Plasmodium (malaria) and Babesia in blood smears. [25] |
| Protargol (Silver Protein) Stain [25] | Impregnates and visualizes the infraciliary lattice and flagellar arrangements of protozoa. | Essential for the precise identification of ciliate and flagellate species based on their unique ciliary/flagellar patterns. [25] |
| Klein's Silver Nitrate Stain [25] | Specifically demonstrates the proteinaceous components of the adhesive disc in mobile peritrich ciliates. | Used for the identification and species determination of trichodinid ciliates in fish. [25] |
| Whole-Slide Scanner [67] | Digitizes entire glass microscope slides at high resolution to create Whole Slide Images (WSIs) for AI analysis. | Generating digital copies of histology slides for input into deep learning models (e.g., using a Hamamatsu NanoZoomer). [67] |
The microscopic identification of intestinal protozoa, long considered the diagnostic gold standard, faces significant challenges in terms of sensitivity, specificity, and the ability to differentiate closely related species [68]. This has prompted a transition toward molecular diagnostic methods, particularly for pathogens like Giardia duodenalis, Cryptosporidium spp., Entamoeba histolytica, and Dientamoeba fragilis [68] [69]. This technical support article frames this transition within the broader context of quality control in protozoan research, providing a comparative analysis of commercial and in-house molecular assays to guide researchers and scientists in selecting, optimizing, and troubleshooting these methods.
Molecular diagnostics offer the potential to overcome the limitations of microscopy, but they introduce new variables that require rigorous quality control, from nucleic acid extraction to final amplification and detection [69]. The following sections provide detailed experimental protocols, troubleshooting guides, and comparative data to support quality assurance in this evolving field.
Experimental Protocols: A Multicenter Comparison
A recent multicenter study provides a robust framework for comparing molecular assays [68]. The methodology below details the key experimental steps.
Sample Collection and Storage
- Sample Type: A total of 355 stool samples were analyzed, comprising 230 freshly collected samples and 125 samples stored in preservation media [68].
- Initial Examination: All samples underwent conventional microscopic examination per WHO and CDC guidelines. Fresh samples were stained with Giemsa, while fixed samples were processed using the formalin-ethyl acetate (FEA) concentration technique [68].
- Storage: Following initial examination, samples were frozen and stored at -20°C until molecular analysis [68].
DNA Extraction Protocol
- Homogenization: A 350 µl volume of Stool Transport and Recovery Buffer (S.T.A.R. Buffer; Roche) was mixed with approximately 1 µl of fecal sample using a sterile loop [68].
- Incubation: The mixture was incubated for 5 minutes at room temperature and then centrifuged at 2000 rpm for 2 minutes [68].
- Supernatant Collection: 250 µl of the supernatant was carefully transferred to a fresh tube and combined with 50 µl of an internal extraction control [68].
- Automated Extraction: DNA was extracted using the MagNA Pure 96 DNA and Viral NA Small Volume Kit on the MagNA Pure 96 System (Roche), a fully automated platform based on magnetic bead separation [68].
PCR Amplification Methods
The study compared two primary RT-PCR methods:
- Commercial Assay: The AusDiagnostics Company RT-PCR test (distributed by Nuclear Laser Medicine) was used according to the manufacturer's instructions [68].
- In-House Assay: A previously validated in-house RT-PCR assay was used. The reaction mixture consisted of:
- 5 µl of extracted DNA
- 12.5 µl of 2× TaqMan Fast Universal PCR Master Mix (Thermo Fisher Scientific)
- 2.5 µl of primer and probe mix
- Sterile water to a final volume of 25 µl [68]
The following diagram illustrates the core comparative workflow of the study:
Comparative Performance Data
The multicenter study yielded quantitative data on the performance of both molecular methods against microscopy and each other for key protozoan parasites [68]. The data below are summarized from the study's findings.
Table 1: Comparative Performance of Molecular Assays for Key Intestinal Protozoa
| Parasite | Commercial vs. In-House PCR Agreement | Key Performance Findings | Notes on Sample Type |
|---|---|---|---|
| Giardia duodenalis | Complete agreement [68] | High sensitivity and specificity, comparable to microscopy [68] | Reliable detection in both fresh and preserved samples [68] |
| Cryptosporidium spp. | High specificity, limited sensitivity for both [68] | Sensitivity limited likely by inadequate DNA extraction from oocysts [68] | — |
| Entamoeba histolytica | — | Molecular assays are critical for accurate diagnosis and differentiation from non-pathogenic species [68] | — |
| Dientamoeba fragilis | High specificity, limited sensitivity for both [68] | Inconsistent detection; sensitivity limited likely by inadequate DNA extraction [68] | — |
| Overall Workflow | — | — | PCR results from preserved stool samples were generally better than from fresh samples, likely due to superior DNA preservation [68] |
Troubleshooting Guide & FAQs
This section addresses common technical issues encountered when working with molecular assays for intestinal protozoa, providing targeted solutions for researchers.
Frequently Asked Questions
Table 2: Frequently Asked Questions on Molecular Assay Implementation
| Question | Evidence-Based Answer & Recommendation |
|---|---|
| Should I replace microscopy with PCR in my lab? | Molecular methods are highly promising, but some authors recommend them as a complement to microscopy, as microscopic examination can reveal additional parasitic infections not targeted by a specific PCR panel [68]. |
| What is the biggest technical challenge in parasite PCR? | The robust wall structure of protozoan cysts and oocysts complicates DNA extraction, often limiting sensitivity. Standardizing and optimizing the DNA extraction procedure is critical for consistent results [68]. |
| How does genetic diversity affect my PCR results? | Interspecific and intraspecific genetic diversity can significantly impact primer and probe binding. Assay design must be based on comprehensive genetic data to ensure detection of all relevant strains [69]. |
| My negative controls show amplification. What should I do? | This indicates contamination. Use new reagent aliquots, especially buffer and polymerase. Ensure use of sterile tips and workstations. "Homemade" polymerases are more prone to contamination; consider using a commercial alternative [70]. |
Troubleshooting Common PCR Problems
Table 3: Troubleshooting Common PCR Issues in Parasite Detection
| Problem | Possible Causes | Recommended Solutions |
|---|---|---|
| No Amplification | - Poor DNA template quality or quantity- Inhibitors in stool sample- Suboptimal primer design or old primers | - Check DNA quality/quantity (e.g., Nanodrop)- Re-purify DNA to remove inhibitors (e.g., with 70% ethanol wash)- Use fresh primer aliquots; verify primer specificity [39] [70] |
| Non-Specific Bands/High Background | - Low annealing temperature- Excess primers, Mg2+, or DNA polymerase- Primer-dimer formation | - Optimize annealing temperature (increase 1-2°C at a time)- Lower primer concentration (0.1–1 µM typical range)- Use hot-start DNA polymerases to increase specificity [39] [70] |
| Low Yield | - Insufficient number of PCR cycles- Low template input- Suboptimal extension time/temperature | - Increase cycle number to 35-40 for low-copy targets- Increase amount of input DNA template- Ensure extension time is sufficient for amplicon length [39] |
| Inconsistent Results Between Replicates | - Pipette calibration errors- Non-homogeneous reagent mixtures- Inhibitors in sample | - Calibrate pipettes regularly- Mix reagent stocks and reactions thoroughly before use- Use fresh, diluted standards and re-purify DNA if needed [70] |
The following decision tree can guide the systematic troubleshooting of a failed PCR reaction:
The Scientist's Toolkit: Research Reagent Solutions
Selecting the appropriate reagents is fundamental to success in molecular parasitology. The table below lists key materials and their functions based on the protocols and troubleshooting advice cited.
Table 4: Essential Research Reagents for Molecular Detection of Intestinal Protozoa
| Reagent / Kit | Specific Function | Research Context & Consideration |
|---|---|---|
| S.T.A.R. Buffer (Roche) | Stool transport and recovery; lyses stool matrix for DNA release [68] | Used in standardized DNA extraction protocols to homogenize samples and protect nucleic acids. |
| MagNA Pure 96 System & Kits (Roche) | Automated, high-throughput nucleic acid extraction using magnetic bead technology [68] | Reduces hands-on time and variability, though manual methods are also common. Critical for overcoming PCR inhibitors from stool. |
| TaqMan Fast Universal PCR Master Mix (Thermo Fisher) | Pre-mixed, optimized solution for real-time PCR, including enzymes, dNTPs, and buffer [68] | Provides a standardized "hot-start" reaction environment, reducing setup time and improving specificity and reproducibility. |
| AusDiagnostics GI Parasite PCR Kit | Commercial multiplex tandem PCR for detection of major intestinal protozoa [68] | Offers a standardized, off-the-shelf solution that minimizes in-house development and validation time. |
| Hot-Start DNA Polymerases | Enzyme engineered to be inactive at room temperature, activated only at high temperatures [39] | Crucial for reducing non-specific amplification and primer-dimer formation during reaction setup, thereby increasing target yield. |
| PCR Additives (e.g., DMSO, GC Enhancer) | Co-solvents that help denature GC-rich DNA and resolve secondary structures [39] | Often essential for amplifying difficult targets, such as those with high GC-content, but require concentration optimization. |
Molecular diagnostics represent a powerful tool for the identification of intestinal protozoa, offering enhanced specificity and the critical ability to differentiate pathogenic from non-pathogenic species [68]. The comparative data shows that both well-validated in-house assays and commercial kits can perform robustly, though challenges in DNA extraction from resilient parasite cysts and oocysts remain a key area for improvement [68].
The future of molecular diagnosis in this field points toward techniques that are more applicable at the point-of-care, such as isothermal amplification methods (e.g., Recombinase Polymerase Amplification or RPA) [71]. Furthermore, the ongoing characterization of genetic diversity in protozoan parasites will be essential for refining primer and probe designs to ensure assays detect all circulating strains [69]. For researchers, a focus on standardized sample collection, nucleic acid extraction, and continuous quality control is paramount for generating reliable, reproducible data that advances both basic science and drug development efforts.
Evaluating Real-World Performance in Prospective Clinical and Research Settings
Troubleshooting Guides and FAQs
Frequently Asked Questions (FAQs)
Q1: What are the most common causes of false-negative results in molecular diagnostics for intestinal protozoa? False negatives in molecular assays are frequently linked to inadequate DNA extraction due to the robust wall structure of protozoan cysts and oocysts, which can resist lysis. One study noted that for targets like Dientamoeba fragilis and Cryptosporidium spp., this can lead to limited sensitivity. Furthermore, the presence of PCR inhibitors in stool samples can also suppress amplification [68].
Q2: In a routine diagnostic workflow, when should microscopy be used alongside multiplex PCR? Microscopy remains essential in the following scenarios:
- When infection with parasites not included in the multiplex PCR panel is suspected (e.g., Cystoisospora belli or helminths), particularly in HIV-infected patients, migrants, or travelers [35].
- To detect non-pathogenic protozoa, which can provide useful ecological context [35].
- As a complementary method when PCR results are inconsistent with clinical presentation, to provide a rapid initial assessment.
Q3: How does sample preservation affect molecular test performance? The method of sample storage is critical for DNA integrity. Studies have demonstrated that results from stool samples preserved in specific media (e.g., Para-Pak) are often superior to those from fresh samples. This is likely because preservatives prevent DNA degradation, leading to more reliable and sensitive PCR results [68].
Q4: What is the clinical significance of detecting Blastocystis spp. and Dientamoeba fragilis with high frequency via PCR? The high frequency of detection for these protozoa with multiplex PCR has prompted investigations into specificity. While their pathogenicity is still debated, their sensitive detection is crucial for high-quality epidemiological studies. Confirmation with simplex qPCR is sometimes used to verify positive results from multiplex assays [35].
Troubleshooting Common Experimental Issues
Problem: Inconsistent results between molecular and microscopic methods.
- Potential Cause: The inherent difference in sensitivity and what each method detects. Microscopy may identify a wider range of parasitic structures, while PCR is more sensitive for specific targeted pathogens but can miss non-targets.
- Solution: Implement an algorithm where microscopy is performed reflexively based on patient epidemiology (travel history, immune status) and clinical symptoms, even when a multiplex PCR is negative for its specific targets [35].
Problem: Low DNA yield from protozoan cysts or oocysts.
- Potential Cause: The chitinous and robust walls of cysts and oocysts are difficult to lyse with standard protocols.
- Solution: Employ a more rigorous lysis method. Recent research on foodborne protozoa has shown that efficient lysis is a prerequisite for sensitive detection. A dedicated lysis device (e.g., OmniLyse) can achieve rapid and efficient disruption within minutes, improving DNA yield significantly for subsequent sequencing or PCR [9].
Problem: High rates of PCR inhibition.
- Potential Cause: Stool samples often contain complex biomolecules that can inhibit polymerase enzymes.
- Solution: The use of an internal extraction control is mandatory to identify inhibition. If inhibition is detected, dilute the DNA template or use a DNA cleanup kit. Automated DNA extraction systems, as used in several studies, can also help standardize the process and reduce inhibition [68] [35].
Summarized Data from Key Studies
Table 1: Detection Rates of Intestinal Protozoa: Molecular vs. Microscopic Methods
This table summarizes key findings from a prospective study analyzing 3,495 stool samples, comparing a commercial multiplex qPCR against traditional microscopy [35].
| Parasite | Detection by Multiplex qPCR | Detection by Microscopy |
|---|---|---|
| Giardia intestinalis | 1.28% (45/3,495) | 0.7% (25/3,495) |
| Cryptosporidium spp. | 0.85% (30/3,495) | 0.23% (8/3,495) |
| Entamoeba histolytica | 0.25% (9/3,495) | 0.68% (24/3,495)* |
| Dientamoeba fragilis | 8.86% (310/3,495) | 0.63% (22/3,495) |
| Blastocystis spp. | 19.25% (673/3,495) | 6.55% (229/3,495) |
Note: Microscopy cannot differentiate the pathogenic *E. histolytica from the non-pathogenic E. dispar, which explains the higher microscopy count [35].*
Table 2: Performance Comparison of Molecular Assays in a Multicentre Study
This table presents data from a multicentre study of 355 samples, comparing a commercial and an in-house RT-PCR against microscopy for specific protozoa [68].
| Parasite | Commercial RT-PCR vs. Microscopy | In-House RT-PCR vs. Microscopy |
|---|---|---|
| Giardia duodenalis | High sensitivity and specificity, complete agreement with in-house PCR | High sensitivity and specificity, complete agreement with commercial PCR |
| Cryptosporidium spp. | High specificity, but limited sensitivity | High specificity, but limited sensitivity |
| Dientamoeba fragilis | High specificity, but inconsistent detection | High specificity, but inconsistent detection |
Detailed Experimental Protocols
Protocol 1: Standardized Workflow for Multicentre Molecular Comparison
This protocol is adapted from a multicentre study evaluating PCR performance [68].
1. Sample Collection and Preparation:
- Collect stool samples (either fresh or preserved in media like Para-Pak).
- For fresh samples, prepare smears and stain with Giemsa for initial microscopy.
- For fixed samples, process using the formalin-ethyl acetate (FEA) concentration technique.
- After microscopic examination, freeze all samples at -20°C for subsequent molecular analysis.
2. DNA Extraction:
- Mix 350 µl of Stool Transport and Recovery Buffer (S.T.A.R. Buffer) with approximately 1 µl of fecal sample using a sterile loop.
- Incubate for 5 minutes at room temperature.
- Centrifuge at 2000 rpm for 2 minutes.
- Carefully collect 250 µl of the supernatant and transfer it to a fresh tube.
- Add 50 µl of an internal extraction control.
- Perform DNA extraction using an automated system (e.g., MagNA Pure 96 System) and a compatible kit (e.g., MagNA Pure 96 DNA and Viral NA Small Volume Kit).
3. In-house Real-Time PCR Amplification:
- Prepare a reaction mixture containing:
- 5 µl of extracted DNA
- 12.5 µl of 2× TaqMan Fast Universal PCR Master Mix
- 2.5 µl of primers and probe mix
- Sterile water to a final volume of 25 µl
- Perform multiplex tandem PCR on a suitable thermocycler (e.g., ABI platform).
Protocol 2: Metagenomic Detection from Leafy Greens
This protocol outlines a novel method for detecting parasites on fresh produce, highlighting an efficient lysis step [9].
1. Sample Spiking and Parasite Recovery:
- Place a 25g lettuce leaf in a sterile container.
- Spike the leaf with a known quantity of parasite oocysts/cysts (e.g., Cryptosporidium parvum, Giardia duodenalis) in 1 ml of PBS, added dropwise over the surface.
- Air-dry for at least 15 minutes.
- Transfer the leaf to a stomacher bag with 40 ml of buffered peptone water with 0.1% Tween.
- Homogenize in a stomacher at 115 rpm for 1 minute.
- Filter the fluid through a 35 µm filter under vacuum to remove plant debris.
- Pellet the oocysts/cysts by centrifuging the filtrate at 15,000x g for 60 minutes at 4°C. Discard the supernatant.
2. Efficient DNA Extraction and Lysis:
- Lyse the pellet containing oocysts/cysts using a dedicated device (e.g., OmniLyse) for 3 minutes to achieve rapid and efficient disruption.
- Precipitate the DNA using acetate precipitation.
- Subject the extracted DNA to whole genome amplification to generate sufficient quantities (e.g., 0.16–8.25 µg) for sequencing.
3. Metagenomic Sequencing and Analysis:
- Prepare libraries from the amplified DNA.
- Perform sequencing using a nanopore device (e.g., MinION) or an alternate platform (e.g., Ion S5 system).
- Upload the generated sequence files (fastq) to a bioinformatic analysis webserver (e.g., CosmosID) for the identification and differentiation of protozoan parasites in the metagenome.
Workflow and Pathway Visualizations
Diagram 1: Diagnostic QC Pathway for Protozoan Identification
Diagram 2: mNGS Workflow for Parasite Detection
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Protozoan Identification Research
| Item Name | Function / Application | Example Use Case |
|---|---|---|
| Stool Transport & Recovery (S.T.A.R.) Buffer | Stabilizes nucleic acids in stool specimens for molecular testing. | Used in DNA extraction protocols prior to automated nucleic acid purification [68]. |
| Multiplex PCR Panels (e.g., AllPlex GIP) | Simultaneously detects multiple protozoan DNA targets from a single sample. | Used for routine, high-throughput screening of common pathogenic protozoa in clinical stools [35]. |
| MagNA Pure 96 System & Kits | Automated, high-throughput nucleic acid extraction system. | Provides standardized, reproducible DNA extraction, reducing human error and cross-contamination [68]. |
| Para-Pak Preservation Media | Preserves parasitic morphology for microscopy and DNA for molecular methods. | Used in multicentre studies to allow for coordinated testing and improve DNA stability compared to fresh samples [68]. |
| OmniLyse Device | Provides rapid and efficient mechanical lysis of robust cyst and oocyst walls. | Critical for efficient DNA recovery from parasites on food surfaces (e.g., lettuce) for metagenomic sequencing [9]. |
| Formalin-ethyl acetate (FEA) | A concentration method for stool samples to enhance microscopic detection. | Used to concentrate parasitic forms from fixed stool samples for microscopic examination [68]. |
Clinical and research laboratories focused on the microscopic identification of protozoan parasites face significant challenges that impact their operational efficiency, testing throughput, and labor requirements. The traditional method for stool parasite testing, the microscopic ova and parasite examination (O&P), is labor-intensive and requires a high level of skill for optimal interpretation, yet remains the cornerstone of diagnostic testing for intestinal protozoa [20]. Laboratories struggle with providing quality O&P results within a clinically significant time frame due to the shortage of skilled technologists capable of reliably evaluating O&P examinations [20]. This technical support center provides troubleshooting guidance and efficiency optimization strategies to address these pressing concerns within the context of quality control for protozoan research.
Troubleshooting Guides
Microscope Performance and Image Quality Issues
Problem: Blurry images or inability to focus across all magnification levels
- Solution: Perform regular calibration using control slides and verify optical alignment [14] [72]
- Preventive Action: Implement weekly visual inspections of all optical components and monthly performance verification with standardized settings [14]
- Expert Tip: "Check for oil on non-oil objectives, which is the top problem encountered. Look through the base into an overhead light to check for damaged or contaminated optics." [72]
Problem: Inconsistent illumination or poor contrast
- Solution: Align the condenser and adjust both aperture and field diaphragms to optimize light path [72]
- Verification: Use control slides to establish baseline illumination settings and document optimal configurations for different specimen types [72]
- Common Pitfall: "With common-use microscopes, condenser settings may be changed by different users. Document optimal iris settings, light intensity, and alignment for consistent results." [72]
Problem: Inaccurate measurements or size determinations
- Solution: Calibrate using micrometer slides and verify measurement accuracy across all objectives [72]
- Quality Control: Maintain calibration records and perform quarterly verification of all measurement systems [14]
Specimen Preparation and Staining Problems
Problem: Low diagnostic yield despite proper collection
- Solution: Ensure adequate specimen volume and proper preservation. Collect multiple specimens (optimally 3) on alternate days to address irregular shedding of protozoa [20]
- Evidence: Studies show evaluating three specimens instead of one increases detection yield by 22.7% for Entamoeba histolytica, 11.3% for Giardia, and 31.1% for Dientamoeba fragilis [20]
Problem: Degraded specimen quality affecting identification
- Solution: Use sodium acetate-acetic acid-formalin (SAF) preservation for long-term storage and maintain consistent storage conditions [21]
- Research Finding: Specimens preserved in SAF can be stored for several months without significant deterioration, maintaining utility for quality assurance programs [21]
Diagnostic Consistency Challenges
Problem: Inconsistent identification results between technologists
- Solution: Implement blinded resubmission programs where selected clinical samples are re-evaluated to assess concordance [21]
- Benchmark Data: Established laboratories achieve approximately 80% concordance rates for pathogenic protozoa when using blinded resubmission assessment tools [21]
- Training Enhancement: Develop affiliations with organizations conducting parasitology surveillance (e.g., CDC DPDx laboratories) to access teaching specimens and maintain technologist proficiency [20]
Frequently Asked Questions (FAQs)
Q: What is the single most impactful efficiency improvement for a parasitology laboratory struggling with turnaround times? A: Implement algorithmic testing that begins with front-line antigen tests for common protozoa like Giardia and Cryptosporidium, reserving traditional microscopic O&P for negative cases or specific clinical indications. This approach significantly reduces labor requirements while maintaining diagnostic accuracy [20].
Q: How can we maintain technologist proficiency with declining positive specimens? A: Establish specimen pooling and sharing agreements with neighboring laboratories, implement regular competency assessment using stored positive specimens, and utilize digital microscopy libraries for continuous training. Positive specimens should be reviewed by all trained technologists to maximize staff competency [20].
Q: What are the key differences in protozoan recovery rates between various produce types? A: Recovery rates vary significantly based on produce characteristics:
Table: Oocyst Recovery Rates from Different Produce Types
| Produce Type | Optimal Processing Method | Average Recovery Rate | Reliable Detection Limit |
|---|---|---|---|
| Berries (general) | Orbital shaking with elution solution | 4.1-12% | 3 oocysts/gram |
| Leafy herbs with soft stems | Stomaching with glycine buffer | 5.1-15.5% | 5 oocysts/gram |
| Aromatic woody-stemmed herbs (e.g., thyme) | Orbital shaking | 5.1-15.5% | 5 oocysts/gram |
| Green onions | Orbital shaking with elution solution | 5.1-15.5% | 5 oocysts/gram |
Source: [73]
Q: How does molecular testing compare with traditional microscopy for efficiency? A: Molecular methods like PCR offer significant efficiency advantages through automation potential and reduced hands-on time, though they require different expertise:
Table: Method Comparison for Protozoan Detection
| Parameter | Traditional Microscopy | Antigen Detection | Molecular Methods (PCR) |
|---|---|---|---|
| Hands-on time | High (15-30 minutes/sample) | Low (<5 minutes/sample) | Medium (varies with automation) |
| Required expertise | Specialized parasitology training | Standard technical training | Molecular biology training |
| Throughput capacity | Low to moderate | High | High with automation |
| Multiplexing capability | Limited | Moderate | High |
| Equipment cost | Low to moderate | Low | High |
Q: What quality control metrics should we track for our microscopy program? A: Implement a comprehensive QC program tracking:
- Concordance rates (target: ≥80% for pathogenic protozoa) [21]
- Specimen adequacy rejection rates
- Turnaround time compliance
- Technologist competency assessment results
- Equipment performance and calibration status [14]
Experimental Protocols for Quality Control
Blinded Resubmission Protocol for Quality Assessment
This protocol assesses diagnostic reproducibility by evaluating concordance between initial and repeated examinations of the same specimens [21].
Materials Needed:
- Sodium acetate-acetic acid-formalin (SAF) preserved stool specimens
- Standard formal-ethyl acetate concentration reagents
- Iron-hematoxylin staining materials
- Microscope with calibrated optics
Methodology:
- Select SAF-preserved clinical stool specimens soon after initial reporting
- Dilute specimens with additional SAF if necessary to create resubmission samples
- Relabel with new accession numbers and fictional patient information
- Introduce blinded specimens into routine workflow
- Compare results from initial and resubmitted examinations
- Calculate concordance rates for targeted protozoan species
Quality Indicator: Concordance rates approximately 80% for pathogenic protozoa represent benchmark performance in established laboratories [21].
Molecular Method Validation Protocol
Materials Needed:
- Commercial or in-house PCR kits
- DNA extraction reagents
- Preserved stool specimens (fresh or in preservation media)
- Real-time PCR instrumentation
Methodology:
- Process 200-300 stool samples using both traditional microscopy and PCR methods
- Use both freshly collected and preserved specimens to account for storage variables
- Extract DNA using optimized protocols for each protozoan species
- Perform parallel testing with microscopy and PCR
- Resolve discrepant results with additional confirmatory testing
Key Consideration: DNA extraction efficiency significantly impacts results, particularly for Cryptosporidium and Dientamoeba fragilis [74]. Preserved stool samples often yield better DNA quality than fresh samples [74].
Workflow Optimization Diagrams
Research Reagent Solutions
Table: Essential Research Reagents for Protozoan Identification
| Reagent/Category | Function/Application | Key Considerations |
|---|---|---|
| Sodium acetate-acetic acid-formalin (SAF) | Specimen preservation for long-term storage | Maintains parasite morphology for months; suitable for quality assurance programs [21] |
| Iron-hematoxylin stain | Permanent staining for enhanced morphological detail | Requires technical expertise but provides superior structural resolution [21] |
| Commercial antigen detection tests (e.g., Remel ProSpecT, TechLab Giardia II) | Rapid screening for specific pathogens | FDA-cleared tests available for Giardia, Cryptosporidium, and Entamoeba histolytica [20] |
| DNA extraction kits optimized for stool samples | Nucleic acid isolation for molecular detection | Critical step affecting PCR sensitivity; particularly important for Cryptosporidium and D. fragilis [74] |
| Fluorescent labels for viability assessment (e.g., Immunofluorescence assays) | Enhanced detection and viability determination | Used in direct fluorescent antibody tests and flow cytometry applications [75] |
| Positive control specimens | Quality assurance and technologist training | Maintain proficiency through shared specimen banks and commercial sources [20] |
Substantial improvements in laboratory efficiency, throughput, and labor utilization are achievable through strategic implementation of algorithmic testing, quality control protocols, and appropriate technology integration. The transition from reliance solely on traditional microscopy to a balanced approach incorporating antigen detection, molecular methods, and well-designed quality assurance programs can address the critical challenges facing modern parasitology laboratories while maintaining diagnostic accuracy essential for quality protozoan research.
Conclusion
The field of quality control for protozoan microscopy is undergoing a profound transformation, moving from a purely manual, expertise-dependent practice to a standardized, technology-driven discipline. The integration of digital microscopy and deep learning, particularly CNNs, demonstrates superior analytical sensitivity and operational efficiency compared to traditional methods, while rigorous validation ensures these tools meet clinical and research standards. Future directions must focus on developing larger, more diverse datasets to improve algorithm robustness, enhancing the accessibility of these technologies in low-resource settings, and fostering a synergistic diagnostic approach that combines AI-powered microscopy with molecular techniques. For researchers and drug development professionals, these advancements are not merely incremental improvements but are pivotal for achieving higher diagnostic precision, accelerating parasitological research, and ultimately improving global health outcomes in the face of evolving parasitic threats.
References
- 1. A Systematic Review and Meta-Analysis, 1999-2024 [https://pubmed.ncbi.nlm.nih.gov/41049918/]
- 2. Parasitic diagnosis: A journey from basic microscopy to cutting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12617590/]
- 3. Global impact of parasitic infections and the importance ... [https://www.frontiersin.org/journals/parasitology/articles/10.3389/fpara.2025.1546195/full]
- 4. Creating and troubleshooting microscopy analysis workflows [https://pmc.ncbi.nlm.nih.gov/articles/PMC10942474/]
- 5. Protein quality control machinery in intracellular protozoan ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6717229/]
- 6. Protein quality control machinery in intracellular protozoan ... [https://pubmed.ncbi.nlm.nih.gov/31228085/]
- 7. Editorial: Recent trends in infection biology and control of ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2023.1292591/full]
- 8. 10 Common Microscope problems [https://microscopemarketplace.com/blogs/news/10-common-microscope-problems?srsltid=AfmBOooAOZW8oRgSgxyEc7GIBAVw6NPNb6NLKYEDhnXOo_8xTCYXvNAr]
- 9. Metagenomic detection of protozoan parasites on leafy ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1566579/full]
- 10. Low-cost, autonomous microscopy using deep learning ... [https://www.sciencedirect.com/science/article/pii/S0952197623011697]
- 11. Depth-enhanced high-throughput microscopy by compact ... [https://www.nature.com/articles/s41467-024-48502-y]
- 12. Safety issues in Microscopy [https://www.microbehunter.com/safety-issues-in-microscopy/]
- 13. Designing a rigorous microscopy experiment: Validating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6504886/]
- 14. Maintaining Quality and Reliability - Light Microscopy Facility [https://lmf.mdibl.org/?page_id=2905]
- 15. An Overview of Techniques for Identification of Parasitic ... [https://www.vin.com/apputil/content/defaultadv1.aspx?pId=11257&id=3864076]
- 16. Diagnostic Methods of Common Intestinal Protozoa [https://pmc.ncbi.nlm.nih.gov/articles/PMC9606991/]
- 17. Real-Time Protozoa Detection from Microscopic Imaging ... [https://www.mdpi.com/2076-3417/14/2/607]
- 18. Deep learning for microscopic examination of protozoan ... [https://www.sciencedirect.com/science/article/pii/S2001037022000423]
- 19. Labeling Strategies to Track Protozoan Parasite Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10895934/]
- 20. Detection of Intestinal Protozoa in the Clinical Laboratory - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3957779/]
- 21. Detection of Pathogenic Protozoa in the Diagnostic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2446938/]
- 22. Intestinal Protozoal Diseases Workup [https://emedicine.medscape.com/article/999282-workup]
- 23. Responses of Protozoan Communities to Multiple ... [https://www.mdpi.com/2076-2615/14/9/1293]
- 24. An Overview of Mucosa-Associated Protozoa: Challenges in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9084232/]
- 25. An Overview of Techniques for Identification of Parasitic ... [https://www.vin.com/apputil/content/defaultadv1.aspx?pId=11257&catId=32250&id=3864076&ind=1536&objTypeID=17]
- 26. 4 Tips for Digital Pathology Slide Scanning [https://www.leicabiosystems.com/us/clinical-solutions/resources/4-tips-for-digital-pathology-slide-scanning/]
- 27. Practical Guidance for Clinical Microbiology Laboratories [https://pmc.ncbi.nlm.nih.gov/articles/PMC5740970/]
- 28. Nets · GitHub Troubleshooting Convolutional Neural [https://gist.github.com/zeyademam/0f60821a0d36ea44eef496633b4430fc]
- 29. [1807.03247] An Intriguing Failing of Convolutional ... Neural Networks [https://arxiv.org/abs/1807.03247]
- 30. Solution to failing Convolutional | Medium neural networks [https://medium.com/the-21st-century/solution-to-failing-convolutional-neural-networks-ff8857b2eaf0]
- 31. c++ - Convolutional not converging - Stack Overflow neural network [https://stackoverflow.com/questions/35190715/convolutional-neural-network-not-converging]
- 32. A metabarcoding approach for detecting protozoan ... [https://www.sciencedirect.com/science/article/abs/pii/S0168160521002749]
- 33. Comparison of DNA quantity and quality from fecal ... [https://www.sciencedirect.com/science/article/pii/S2352771424000570]
- 34. Comparative analysis of commercial and “In-House ... [https://parasitesandvectors.biomedcentral.com/articles/10.1186/s13071-025-06879-9]
- 35. Improvement of the diagnosis of intestinal protozoa using a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12077082/]
- 36. Tannic Acid in TEM of Parasitic Protozoa - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12583016/]
- 37. PCR Troubleshooting: Common Problems and Solutions [https://www.mybiosource.com/learn/pcr-troubleshooting-common-problems-and-solutions/]
- 38. PCR Troubleshooting Guide [https://www.genscript.com/pcr-troubleshooting-guide.html]
- 39. PCR Troubleshooting Guide | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-troubleshooting.html]
- 40. Polymerase Chain Reaction: Basic Protocol Plus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4846334/]
- 41. How to Troubleshoot Sequencing Preparation Errors (NGS ... [https://www.cd-genomics.com/resource-sequencing-preparation-troubleshooting.html]
- 42. Ten common issues with reference sequence databases ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2024.1278228/full]
- 43. Ten common issues with reference sequence databases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10978663/]
- 44. The effects of sequencing strategies on Metagenomic ... [https://www.sciencedirect.com/science/article/pii/S240584402409460X]
- 45. Deep learning-driven automated high-content dSTORM ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11986538/]
- 46. Harmonization of Protocols for Multi-Species Organoid ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2020.610368/full]
- 47. CDC - DPDx - Diagnostic Procedures - Stool Specimens [https://www.cdc.gov/dpdx/diagnosticprocedures/stool/specimencoll.html]
- 48. Parasitology Specimen Collection Guidelines [https://www.laboratoryalliance.com/healthcare-providers/laboratory-services/specimen-collection-documents/parasitology-specimen-collection-guidelines/]
- 49. TEM Fixation - Protocols - Microscopy [https://biotech.unl.edu/tem-fixation-protocols-microscopy/]
- 50. DNA Extraction from Protozoan Oocysts/Cysts in Feces for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4096637/]
- 51. Evaluation of Three Protocols of DNA Extraction for ... [https://brieflands.com/journals/jjm/articles/63096]
- 52. Molecular Biology Can Change the Classic Laboratory ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2017.02191/full]
- 53. Validation of Digital Slide Scanning and a Convolutional ... [https://www.mdpi.com/2075-4418/15/23/2974]
- 54. Improving the accuracy of automated labeling of specimen ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12629457/]
- 55. Detection of protozoan and helminth parasites in concentrated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12607898/]
- 56. Optical Contrast Methods | Learn & Share [https://www.leica-microsystems.com/science-lab/microscopy-basics/optical-contrast-methods/]
- 57. Microscopy Basics | Enhancing Contrast in Transmitted Light [https://zeiss-campus.magnet.fsu.edu/articles/basics/contrast.html]
- 58. Contrast Enhancement Technique MTF Curves [https://evidentscientific.com/en/microscope-resource/tutorials/mtf/contrasttechniques]
- 59. Deep learning for microscopic examination of protozoan ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8886013/]
- 60. AI in radiology: From promise to practice − A guide ... [https://www.sciencedirect.com/science/article/abs/pii/S0720048X2400514X]
- 61. Development of AI-assisted microscopy frameworks ... [https://www.nature.com/articles/s42256-024-00903-w]
- 62. Modern Ways to Monitor Microscope Performance [https://evidentscientific.com/en/insights/modern-ways-to-monitor-microscope-performance-from-built-in-to-external-tools]
- 63. Where Have All the Diagnostic Morphological Parasitologists ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9667774/]
- 64. Contrast in Optical Microscopy [https://evidentscientific.com/en/microscope-resource/knowledge-hub/techniques/contrast]
- 65. Performance validation of the BD MAX Enteric Parasite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11895086/]
- 66. Performance validation of deep-learning-based approach in ... [https://parasitesandvectors.biomedcentral.com/articles/10.1186/s13071-025-06878-w]
- 67. Artificial intelligence-assisted diagnosis of glomerular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12538914/]
- 68. Comparative analysis of commercial and “In-House ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12317615/]
- 69. The impact of genetic diversity in protozoa on molecular ... [https://www.sciencedirect.com/science/article/abs/pii/S1471492210002345]
- 70. PCR Troubleshooting Guide & Solutions [https://www.bosterbio.com/protocol-and-troubleshooting/pcr-troubleshooting?srsltid=AfmBOoquFTrmaw1yhM68uoW9cY9SMDVb8bU9qXfBEPIEO7Y8ZC6JD8Ci]
- 71. Molecular diagnosis of protozoan parasites by Recombinase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6779323/]
- 72. How to Troubleshoot Laboratory Microscopes: A Guide [https://www.linkedin.com/advice/0/what-most-important-steps-take-when-troubleshooting]
- 73. Optimization and validation of methods for isolation and real-time PCR identification of protozoan oocysts on leafy green vegetables and berry fruits - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S2405676615300299]
- 74. Comparative analysis of commercial and "In-House ... [https://pubmed.ncbi.nlm.nih.gov/40751255/]
- 75. Waterborne protozoan parasite detection using two-frequency ... [https://pubs.rsc.org/en/content/articlehtml/2025/ay/d5ay00184f]